RESEARCH ARTICLE
Formulation and Pharmaceutical Evaluation of Capsules Containing the n-Hexane Fraction of Moringa oleifera Leaves
Academic Editor: Bharath Kumar Chagaleti
Sciences of Phytochemistry|Vol. 5, Issue 1, pp. 105-121 (2026)
CC BY 4.0-2026 Authors
Views
Downloads
Shares
Received
Jan 21, 2026Revised
Mar 11, 2026Accepted
Apr 3, 2026Published
Apr 6, 2026
Abstract
This study investigated the phytochemical composition, safety profile, and pharmaceutical formulation of capsules containing the hexane fraction of Moringa oleifera leaves (MOHx). The main objective of this study was to comprehensively evaluate the phytochemical constituents, toxicity profile, and develop a standardized oral capsule formulation of the hexane fraction of M. oleifera leaves suitable for potential therapeutic application. Phytochemical profiling was performed using LC–MS and GC–MS, and compounds were tentatively identified based on spectral library matching and fragmentation patterns. Selected phytochemicals were evaluated using in silico ADMET prediction and molecular docking analyses. Acute oral toxicity was assessed in Wistar rats using Lorke’s method. Pre-formulation studies were conducted prior to capsule formulation using the wet granulation technique. The granules were evaluated for micromeritic properties, and the capsules were subjected to pharmacopoeial quality tests including weight uniformity, disintegration, and dissolution. The estimated LD₅₀ of the extract was 3.808 mg/kg body weight, indicating relatively low acute toxicity. Dissolution testing showed more than 80% release within 20 min under the experimental conditions employed. These findings suggest that the developed capsule formulation provides a suitable pharmaceutical dosage form for the hexane fraction of M. oleifera, although further studies including stability testing, quantitative phytochemical standardization, and pharmacological evaluation are required.
Introduction
Herbal medicine remains an integral component of healthcare delivery in Nigeria, deeply rooted in cultural heritage and traditional healing practices (1). Among medicinal plants of significant interest, Moringa oleifera Lam. (family: Moringaceae), commonly referred to as the “miracle tree,” has gained considerable scientific and commercial attention due to its diverse nutritional and pharmacological properties. Although native to South Asia, the plant is widely cultivated across Africa, including Nigeria, where it is used for both dietary and therapeutic purposes (2). Various parts of the plant, including the leaves, seeds, pods, flowers, bark, and roots, have traditionally been employed in the management of conditions such as inflammation, infections, malnutrition, hypertension, and diabetes (3).
Phytochemical investigations have revealed that M. oleifera contains a wide array of bioactive constituents, including alkaloids, flavonoids, tannins, saponins, terpenoids, and essential fatty acids, which contribute to its broad pharmacological profile (4, 5). While most studies have focused on aqueous and ethanolic extracts due to their richness in polar phenolic compounds, increasing attention has been directed toward lipophilic fractions, particularly those obtained using n-hexane. These non-polar fractions are enriched with compounds such as tocopherols, β-sitosterol, campesterol, and unsaturated fatty acids, including oleic and linoleic acids, which have been associated with antioxidant, anti-inflammatory, antiapoptotic, and antimicrobial activities (6–8). Notably, emerging evidence from our ongoing bioassay-guided studies involving M. oleifera and Psidium guajava leaf extracts has demonstrated that the hexane fraction of M. oleifera exhibited the most pronounced antiviral activity in an in ovo model against Newcastle disease virus (NDV). Although detailed findings are yet to be fully reported, this preliminary observation provides a biological basis for further investigation and informed the selection of the hexane fraction (MOHx) for the present study.
Despite the growing evidence supporting the therapeutic potential of MOHx, its practical application remains limited by formulation challenges associated with lipophilic phytoconstituents. These include poor aqueous solubility, susceptibility to oxidative degradation, and low oral bioavailability (9). Furthermore, the lack of standardization in herbal preparations often results in variability in dosage, efficacy, and reproducibility of therapeutic outcomes (10). Addressing these limitations necessitates the development of appropriate pharmaceutical dosage forms capable of enhancing stability, ensuring dose uniformity, and improving patient compliance.
Recent advances in phytopharmaceutical development have emphasized the formulation of standardized solid dosage forms for plant-derived products (11, 12). Capsule formulations, in particular, offer advantages such as accurate dosing, improved stability, ease of administration, and enhanced patient acceptability (11–13). The incorporation of suitable pharmaceutical excipients further improves flow properties, compressibility, disintegration, and overall performance of formulations, especially for lipophilic extracts such as MOHx (14).
In this context, the present study was designed to systematically develop and comprehensively evaluate a standardized herbal capsule formulation containing the hexane fraction of Moringa oleifera (MOHx). The study involved the extraction of the lipophilic fraction, granule preparation, and detailed evaluation of pre-compression parameters, including flowability and compressibility. Post-encapsulation assessments included weight uniformity, disintegration time, and in vitro dissolution profiling in accordance with established pharmacopoeial guidelines.
To support the formulation rationale, comprehensive phytochemical characterization of the hexane fraction was performed using liquid chromatography–mass spectrometry (LC–MS) and gas chromatography–mass spectrometry (GC–MS). Selected compounds identified from the extract were further systematically subjected to in silico absorption, distribution, metabolism, excretion, and toxicity (ADMET) prediction, as well as molecular docking studies, to explore their potential biological interactions. These computational analyses provide valuable and supportive molecular-level insights into the pharmacological relevance of the identified constituents. Overall, this study integrates phytochemical profiling, computational prediction, and pharmaceutical formulation approaches to successfully develop a standardized oral capsule dosage form of MOHx. This integrated strategy aims to significantly enhance the stability, handling, and dosing accuracy of the lipophilic extract, thereby contributing to the advancement of evidence-based phytopharmaceutical development.
Experimental Section
Materials
All reagents and solvents used in this study were of analytical grade. Materials included hard gelatin capsule shells, lactose (diluent), microcrystalline cellulose (disintegrant), starch (binder), magnesium stearate (lubricant), ethanol (95% v/v), n-hexane, phosphate buffer solutions, 0.1 N HCl, 0.1 N NaOH, and distilled water, capsulation and, UV machine.
Plant Material Collection and Authentication
Fresh leaves of M. oleifera were collected from Wamako LGA, Sokoto State, Nigeria (coordinates: 13.10°N, 5.15°E) in September 2021. The plant wasidentified and authenticated by a taxonomist at the Department of Pharmacognosy and Ethnopharmacy, Usmanu Danfodiyo University, Sokoto, and avoucher specimen (No. PCG/UDUS/Mor/001) was deposited in the departmental herbarium for future reference. The leaves were washed, shade-dried at room temperature (28 ± 2 °C) for two weeks, and pulverized into a fine powder.
Preparation of the Hexane Fraction (MOHx)
The powdered leaves (1 kg) were subjected to maceration in n-hexane using a plant-to-solvent ratio of 1: 4 (w/v) for 48 h at room temperature with intermittent agitation. The extraction was performed in triplicate (n = 3) to ensure reproducibility, and the percentage yield was expressed as mean ± standard deviation. The mixture was filtered using stainless steel mesh (100–200 µm) followed by Whatman No. 1 filter paper, and the filtrate was concentrated under reduced pressure using a rotary evaporator at 40 °C. The extract was further dried in a desiccator to obtain a semi-solid mass (MOHx), which was stored in an airtight container at 4 °C. Thus it should be noted that, residual solvent removal was achieved by rotary evaporation under reduced pressure and further drying in the desiccator; however, quantitative residual solvent analysis was not performed and is acknowledged as a limitation.
Phytochemical Screening
Preliminary qualitative phytochemical screening of the hexane fraction of M. oleifera (MOHx) was carried out using standard procedures as described in literature (15-17). The screening was conducted as a preliminary and exploratory assessment of the chemical classes present in the extract.
Given the non-polar nature of the hexane fraction, it is important to note that conventional qualitative colorimetric tests are primarily designed for polar metabolites; therefore, the results obtained should be interpreted with caution and regarded as indicative rather than definitive.
Liquid Chromatography-Mass Spectrometry (LC-MS) Conditions
The extract was prepared by dissolving an appropriate quantity of MOHx in methanol and filtering through a 0.45 µm membrane filter prior to analysis. Chromatographic separation was achieved using a reverse-phase C18 column (Sunfire C18, 4.6 × 150 mm, 5 µm particle size). The mobile phase consisted of water containing 0.1% formic acid (solvent A) and acetonitrile (solvent B) delivered in a gradient mode at a flow rate of 1.0 mL/min. Mass spectrometric detection was performed using electrospray ionization (ESI) in both positive and negative ionization modes, with a scan range of m/z 100–1250. Compound identification was based on comparison of mass spectral data with available spectral libraries and interpretation of fragmentation patterns. Accordingly, all identified compounds are reported as tentatively identified. The detection of certain polar compounds within the hexane fraction is interpreted cautiously and may reflect trace carryover, matrix effects, or analytical artefacts rather than true selective extraction into the non-polar solvent. Data were processed in Empower 3 software (18).
Gas Chromatography-Mass Spectrometry (GC-MS) Conditions
GC–MS analysis was performed using a gas chromatograph coupled with a mass spectrometer (e.g., PerkinElmer Clarus 500 system) equipped with an Elite-5MS capillary column (30 m × 0.25 mm internal diameter × 0.25 µm film thickness). Helium was used as the carrier gas at a constant flow rate of 1.0 – 1.2 mL/min. The oven temperature was programmed from 70 °C (held for 2 min) to 300 °C at a ramp rate of 10 °C/min, with a final hold time of 5–10 min. Mass spectra were acquired over a scan range of m/z 35–650. Compound identification was performed by comparison of the obtained spectra with those in the NIST mass spectral library, using a similarity index of ≥90% as the acceptance criterion. However, retention indices and authentic reference standards were not employed; therefore, compound identification should be considered tentative. The occurrence of methyl ester derivatives in the chromatogram may be attributed to in situ formation during analysis, possible transesterification reactions, or limitations inherent in library-based matching, and thus should be interpreted with caution.
In vivo Acute Toxicity Studies
Adult Wistar rats (weighing 120–150 g, both sexes) were sourced from the Ahmadu Bello University, Zaria, Nigeria. The animals were acclimatized in the animal facility at the Faculty of Pharmaceutical Sciences, Usmanu Danfodiyo University, Sokoto, under standard laboratory conditions (12-hour light/dark cycle, temperature maintained at 23 ± 3 °C), with unrestricted access to water and pellet diet. The study protocol was approved by protocol was approved by the Health Research and Ethics Committee (HREC) of Usmanu Danfodiyo University, Sokoto-Nigeria (Reference No: UDUS/UREC/2020/006; Date: October 20, 2020). Acute oral toxicity of MOHx was evaluated using the method of Lorke (19), which involves a two-phase experimental design. In the initial phase, nine rats of both sexes were randomly assigned into three groups (n = 3 per group) and administered doses of 10, 100, and 1000 mg/kg body weight, respectively. The animals were monitored for 24 h for signs of toxicity and mortality. In the second phase, four animals (one per dose group) received higher doses of 1200, 1600, 2900, and 5000 mg/kg body weight, respectively. The extract was administered at a uniform volume of 10 mL/kg, prepared as a suspension in 2% (v/v) Tween 80 to ensure consistent dosing. The median lethal dose (LD₅₀) was subsequently estimated based on the observed outcomes.
In Silico Studies
Molecular Properties and Drug-likeness of the Ligands
The Swiss ADME tool was used to calculate the molecular properties of the ligands (20). The molecular properties were screened based on the “Lipinski's rule of five (21). The molecular weight, number of rotatable bonds, number of hydrogen bond donors and acceptors, molar refractivity, total polar surface area (TPSA) and the partition coefficient between n-octanol and water (log P o/w) were determined (20).
Pharmacokinetic Profile
SwissADME server was used to determine the pharmacokinetic profile of the phytochemicals. The parameters determined include; Blood brain barrier (BBB) permeation, Human Intestinal Absorption (HIA), P-glycoprotein substrate (P-gp), cytochrome P450 isoform inhibition, Skin permeation Log Kp and bioavailability score (20).
Protein Preparation and Ligand Preparation
The three-dimensional (3D) structure of the Newcastle disease virus (NDV) hemagglutinin–neuraminidase (HN) protein (PDB ID: 1E8U) (22) was retrieved from the Protein Data Bank (PDB), given its critical role in viral attachment and entry into host cells. Protein preparation involved removal of co-crystallized ligands and water molecules, addition of polar hydrogen atoms, and energy minimization to obtain a stable conformation suitable for docking analysis (23).
The structures of selected compounds (7, 8-Dihydropteroic acid, Jasmonic acid, Citreoisocourmarin, 2-furoic acid, Linoleic acid, Oleic acid) were obtained from the PubChem database in SDF format and subjected to geometry optimization prior to docking (24).
Molecular Docking Studies
Molecular docking studies were performed to evaluate the potential interactions between selected phytoconstituents identified from MOHx and a relevant viral target associated with antiviral activity. Docking simulations were performed using AutoDock Vina implemented in PyRx 0.9.x. (24) The grid box was defined to encompass the active binding site of the hemagglutinin–neuraminidase protein, with center coordinates set at x = 25.0, y = 35.0, and z = 15.0, and dimensions of 25 × 25 × 25 Å. These parameters were selected to ensure adequate coverage of the binding pocket while allowing sufficient flexibility for ligand conformations during docking. The grid settings were determined based on the reported active site region of the protein and established docking practices. No site-directed docking validation (e.g., redocking of a co-crystallized ligand) was performed, and this is acknowledged as a limitation of the study.
The selection of NDV as a docking target is supported by prior preliminary antiviral findings, as previously introduced and discussed earlier within the study. Importantly, the docking results are presented as supportive computational predictions intended to provide additional insight into possible molecular interactions and binding behavior, and were not used to establish or confirm any biological or antiviral activity.
Toxicity Prediction
In silico toxicity prediction of selected phytoconstituents was performed using the Protox-II web server, with molecular structures retrieved from the PubChem database (25). Predicted endpoints included median lethal dose (LD₅₀), toxicity class, hepatotoxicity, cytotoxicity, immunotoxicity, and mutagenicity, based on established computational models (25). The predicted toxicity parameters were not directly equated with experimentally determined LD₅₀ values obtained from in vivo acute toxicity studies, as both approaches are based on fundamentally different models and assumptions. Differences observed between predicted and experimental LD₅₀ values were interpreted as expected variations between computational estimations and biological responses, rather than inconsistencies. Furthermore, confidence intervals and applicability domain metrics of the predictive models were not explicitly provided by the software and are acknowledged as inherent limitations of the computational approach. Therefore, the in silico toxicity results are presented as complementary estimates to support preliminary safety evaluation and should not be considered a substitute for experimental toxicological assessment.
Pre-formulation Study of MOHx
Pre-formulation studies were conducted to evaluate the physicochemical properties of the hexane fraction of MOHx and to provide a basis for formulation into capsule dosage form.
Determination of pH
The pH meter was calibrated with two buffer solutions. 20 mL of water was added to a 20 g sample in a beaker, covered, and stirred for 5 min. The pH of the sample was taken after the mixture stood for 15 min (26, 27).
Solubility Test
The solubility of MOHx was investigated in selected solvents, including distilled water, ethanol (95%), HCl (0.1N), NaOH (0.1N), NaCl (0.9%), and phosphate buffer (pH 6.8), using a mechanical shaking method. An equilibrium solubility approach was employed by adding an excess amount of the test sample, MOHx to10 mL of each solvent, followed by continuous shaking for 24 h at 25 °C to attain saturation equilibrium. The mixtures were subsequently filtered, and the filtrates analyzed. However, quantitative determination of saturation solubility (mg/mL) was not fully established due to the absence of validated analytical calibration data, and therefore the results are interpreted as semi-quantitative. This is acknowledged as a limitation of the study (26).
UV–Visible Spectrophotometric Analysis of MOHx and Calibration Curve
The maximum absorption wavelength (λmax) of MOHx was determined using a UV–Visible spectrophotometer following the procedure described by Sneha et al with slight modification. Briefly, a stock concentration of the extract was prepared by weighing 0.5 mL of the extract and making it up to volume using 95% ethanol. The prepared sample was then scanned between 450-200 nm against ethanol as blank (26).
Preparation of the Calibration Curve
A series of standard solutions of the extract were prepared in an appropriate solvent (dilutions of five different concentrations ranging from 0.5 to 3 mg/mL were prepared and volume made up using ethanol), and absorbance of these dilutions was measured using UV-visible spectrophotometer at selected wavelength against ethanol as blank; the absorbance values were recorded to generate a calibration curve (supplementary file). Although a calibration curve was constructed for analytical estimation, detailed method validation parameters, including precision, accuracy, and robustness, were not comprehensively evaluated. These limitations are acknowledged and warrant further validation in future studies.
Formulation Granules
Four batches of granules containing the hexane fraction of M. oleifera (MOHX) were formulated using the wet granulation technique (28), with each batch designed to yield approximately 150 capsules. The previously obtained dried extract (7.5 g per batch) was blended with lactose (18.0 g), which served as a diluent to improve bulk and handling properties (28, 29). Starch was incorporated as a binder in varying concentrations across the four batches (15.0 g, 11.25 g, 7.5 g, and 3.75 g), while microcrystalline cellulose was included as a disintegrant in corresponding complementary proportions (3.75 g, 7.5 g, 11.25 g, and 15.0 g), respectively. Magnesium stearate (0.09 g) was uniformly added as a lubricant in all formulations (28). The selection and proportions of excipients were informed by preliminary pre-formulation studies, with the aim of achieving acceptable flow properties, compressibility, and capsule integrity (28, 30). However, no formal experimental optimization design (e.g., factorial or response surface methodology) was employed to systematically justify the excipient ratios, and this is acknowledged as a limitation of the formulation development process. Granulation was carried out using ethanol (95% v/v) as the granulating fluid, added in sufficient quantity to produce a cohesive wet mass. The resulting mass was passed through an appropriate sieve to form granules, which were then evenly spread on stainless steel trays and dried in a hot air oven at 45 °C for 48 h (28). Following the initial drying, the granules were resized by passing them through a No. 22 sieve (0.8 mm) and subjected to further drying at 45 °C for an additional 24 h to achieve constant weight (28). Although drying was continued until a constant weight was obtained, the residual moisture content was not quantitatively determined using standard methods (e.g., loss on drying), which is recognized as a limitation (26, 27). The final dried granules were appropriately labeled and stored in airtight containers pending further evaluation and encapsulation.
Evaluation of the Granules (Micromeritic Properties)
The prepared granules were evaluated for micromeritic properties, including angle of repose, tapped density, bulk density, Carr’s index, and Hausner ratio using standard procedures (28-30). These parameters were used to assess the flow characteristics and suitability of the granules for capsule filling.
Determination of Angle of Repose
A stainless-steel funnel with a 10 mm orifice diameter and 111 mm length from the top to the end of the orifice was fixed at 4 cm from the bench to the funnel orifice. 5 g of powder sample was charged into the funnel and allowed to fall. The height (h) and width (w) of the pile were measured. The results were considered valid only when a symmetric cone was obtained. The procedure was repeated in triplicate. Angle of repose (θ) was calculated using Equation 1.
Determination of Bulk Density and Tapped Density
For the bulk density, 10 g sample was transferred into a 25 mL graduated cylinder with 0.5 mL mark. The cylinder was manually tapped gently twice on a table top surface, and the volume was measured. Bulk density was calculated using Equation 2.
Where w is the weight of the sample, and vb is the volume occupied by the bulk powder.
For the tapped densitythe cylinder was then fixed to the tap density tester and subjected to 500, 750 and 1250 taps at a rate of 250 taps/min. After each specified number of taps, the tapped volume was measured. Tapped density was calculated using Equation 3.
Where w is the weight of the sample, and vt is the volume occupied by the powder after tapping.
Determination of Hausner's ratio and carr's compressibility index
The results of bulk and tapped densities were used to calculate Carr’s compressibility index and Hausner’s ratio. These parameters help in estimating the flow properties and compressibility of the powder (28, 30). The formulae are given below for the Hausner’s ratio (Equation 4) and the Carr’s compressibility index (Equation 5).
Formulation of the Capsules
The encapsulation machine was thoroughly cleaned and dried prior to use. Empty hard gelatin capsules were placed on the removable plate, ensuring that the capsule bodies were oriented downward. The caps were carefully separated from the bodies. The capsule bodies were then filled with the previously prepared granules from each of the four batches. After filling, the capsule shells were reassembled and the filled capsules were ejected from the machine. This procedure was repeated for all batches of the prepared granules. Following encapsulation, the formulated capsules were subjected to quality control evaluation. These included weight uniformity, disintegration time, and in vitro dissolution testing, performed in accordance with standard pharmacopeial methods (26-27). However, content uniformity testing, which is essential for ensuring uniform distribution of active constituents in capsule dosage forms, was not conducted in this study and is acknowledged as a limitation.
Characterization of the Prepared Capsules
Weight Uniformity Test
The uniformity of capsule weight was evaluated in accordance with standard pharmacopoeial guidelines (USP/BP) (26, 27). Twenty capsules were randomly selected from each batch and individually weighed using a calibrated analytical balance. The contents of each capsule were carefully removed, and the empty shells were weighed. The net fill weight of each capsule was calculated by subtracting the shell weight from the total capsule weight. The mean weight, standard deviation, and percentage deviation of individual capsule weights from the mean were calculated to assess compliance with pharmacopoeial limits.
Disintegration Test
The disintegration test was carried out using a USP disintegration apparatus. Six capsules from each batch were randomly selected and placed individually into the tubes of the apparatus. The test was conducted using simulated saliva fluid (pH 6.8) as the disintegration medium, maintained at 37 ± 0.5 °C. The apparatus was operated according to standard pharmacopoeial conditions (27), and the time required for each capsule to disintegrate completely into particles capable of passing through a 10-mesh screen was recorded.
Dissolution Test
In vitro dissolution studies were performed using the USP Dissolution Apparatus II (paddle method). The dissolution medium consisted of 900 mL of 0.1 N hydrochloric acid, maintained at 37 ± 0.5 °C to simulate physiological conditions. The paddle rotation speed was set at 100 rpm. Capsules equivalent to the predetermined drug content were introduced into the dissolution vessels. At predetermined time intervals (10, 20, 30, 40, 50, and 60 min), 10 mL samples were withdrawn using a syringe and immediately replaced with an equal volume of fresh dissolution medium maintained at the same temperature to ensure sink conditions. The withdrawn samples were filtered and analyzed spectrophotometrically at the appropriate wavelength. The cumulative percentage drug release was calculated and plotted against time in accordance with standard pharmacopeial procedures (26, 27).
Results and Discussion
Extraction and Phytochemical Profile of MOHx
The MOHx was olive-green coloured semi-solid and weighed 10.0 g with a percentage yield of 1%. The phytochemical screening revealed the presence of metabolites of tannins, phenolic and, steroids, etc. , as presented in Table 1.
| Phytochemicals Tested | Intensity of Reaction (Detection) | ||
|---|---|---|---|
| Low | Medium | High | |
| Alkaloid | - | - | - |
| Tannins | ++ | ||
| Flavonoids | - | - | - |
| Phenolics | ++ | ||
| Saponins | - | - | - |
| Steroids | ++ | ||
| Glycosides | - | - | - |
| Reducing Sugars | - | - | - |
| Key: + Low ++Medium +++ High; -not detected. | |||
LCMS Chromatographic Profiling and Tentative Identification of Compounds from MOHx
LC–MS profiling of MOHx was performed using a triple-quadrupole instrument in both positive and negative electrospray ionization (ESI) modes, with data processed using Empower 3 software. In the positive ionization mode, the total ion chromatogram (TIC) exhibited approximately 36 detectable peaks over a retention time range of 0.05–19.7 min, with signal intensities ranging from ~3.000 to > 12.000 counts.
Thus, as presented in Table 2, preliminary compound assignments were made using database matching (METLIN, PubChem, and HMDB) (31, 32) combined with interpretation of precursor ion m/z values and MS/MS fragmentation patterns. A subset of peaks was tentatively identified as 2-furoic acid, jasmonic acid, citreoisocoumarin, a 7-O-methylated flavonoid, 7, 8-dihydropteroic acid, and linoleic acid. These assignments were supported by consistency in molecular weights and fragmentation behavior, as presented in Figure 1 and Supplementary Data.
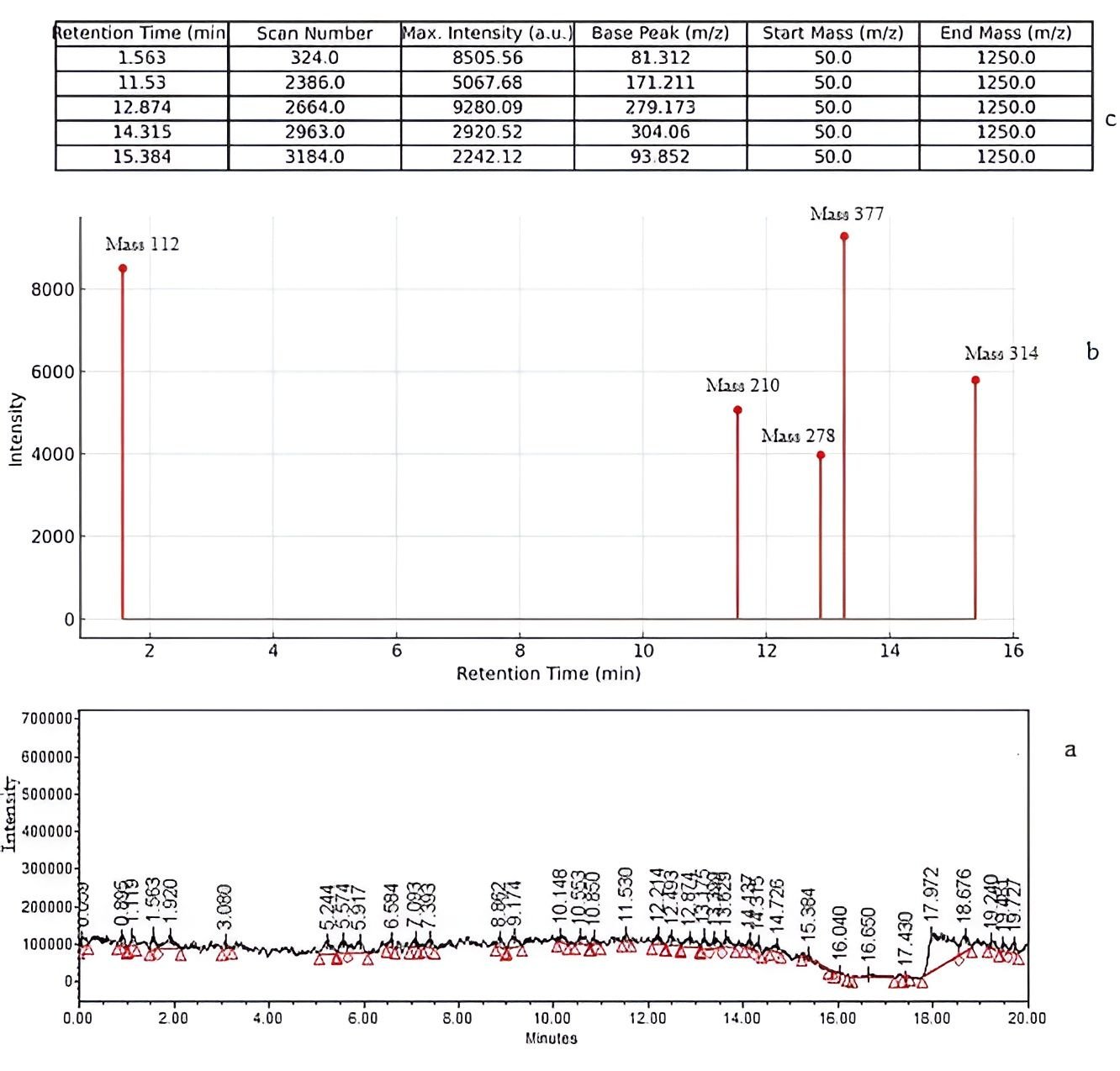
However, it is important to emphasize that these identifications remain tentative, as no confirmation with authentic reference standards or validated spectral libraries was performed, which is a recognized limitation in untargeted LC–MS profiling of complex plant matrices (33). Furthermore, quantitative parameters such as peak area normalization, relative abundance, and confidence scoring were not established, limiting the reliability and reproducibility of compound characterization.
Notably, the detection of relatively polar compounds (e.g., 7, 8-dihydropteroic acid) in a non-polar hexane fraction is unexpected and may reflect trace co-extraction, matrix carryover, or ionization artifacts, phenomena that have been reported in LC–MS analyses of plant extracts when using broad ionization conditions (34). Additionally, the detection of fatty acids such as linoleic acid in positive ESI mode without derivatization should be interpreted cautiously, as ionization efficiency for such compounds is typically limited under these conditions (34, 35).
Overall, while LC–MS provides valuable preliminary insight into the chemical profile of MOHx, the absence of method validation and confirmatory analyses necessitates cautious interpretation of these findings.
| S/n | Rt Time (min) | Max Intensity | Av MW (g/mol) | Precur-sor Ion (m/z) | Product Ion (s) (m/z) | Ionization Mode | Molecular Formula | Compound Name |
|---|---|---|---|---|---|---|---|---|
| 1 | 1.563 | 8505.557031 | 112 | 113 | 85, 69, 57 | QqQ/ESI (+) | C5H4O3 | 2-Furoic acid |
| 2 | 11.530 | 5067.676710 | 210 | 211 | 193, 165, 147 | QqQ/ESI (+) | C12H18O3 | Jasmonic acid |
| 3 | 12.874 | 9280.084990 | 278 | 279 | 261, 243, 215 | QqQ/ESI (+) | C15H10O5 | Citreoisocoumarin |
| 4 | 14.315 | 2920.518750 | 377 | 378 | 360, 345, 330 | QqQ/ESI (+) | C18H16O8 | 2-(3, 4-dihydroxyphenyl)-5-hydroxy-3, 6, 7-trimethoxy-chromen-4-one (flavonoid) |
| 5 | 15.384 | 2242.148750 | 314 | 315 | 297, 280, 252 | QqQ/ESI (+) | C7H10N5O3S | 7, 8-Dihydropteroic acid |
| 6 | 12.473 | 9280.084990 | 280 | 279 | 261, 241, 223 | QqQ/ESI (−) | C18H32O2 | *Linoleic acid |
| Note: Retention times were matched from the TIC and summary MS table (Figure 1 and Table 2); *Linoleic acid was predicted but not observed in the positive ion mode chromatogram; LC-QqQ-MS = Liquid Chromatography - Triple Quadrupole Mass Spectrometry. | ||||||||
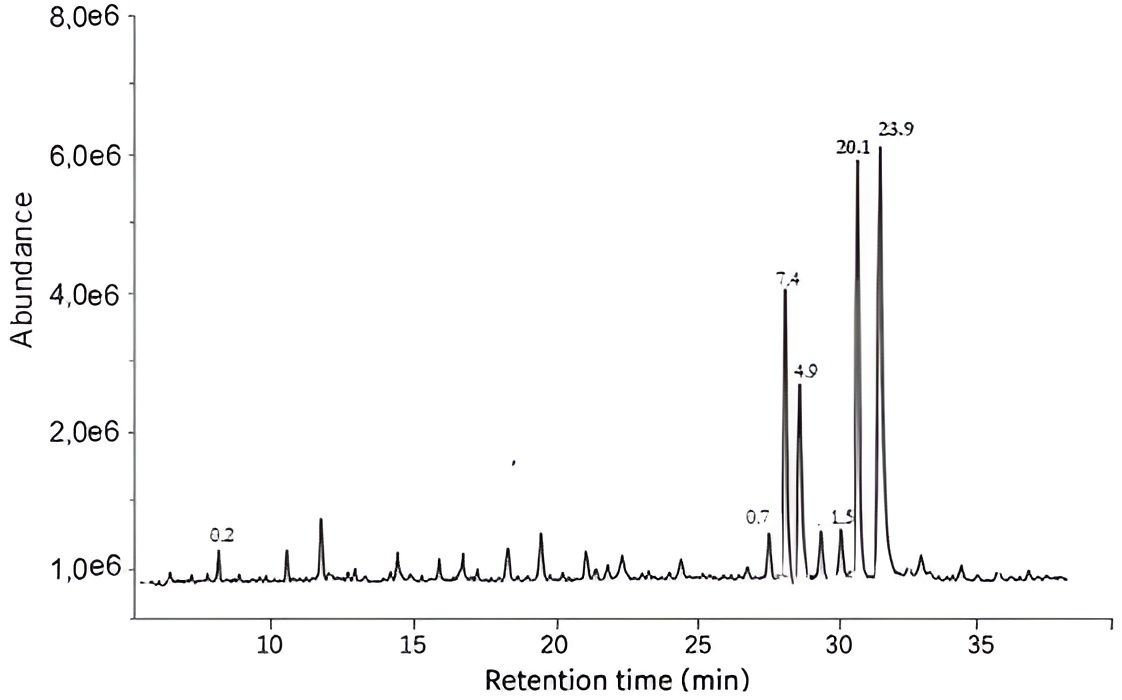
GC–MS Analysis and Identification of Lipophilic Constituents of MOHx
Complementary GC–MS analysis of MOHx revealed a profile dominated by lipophilic constituents (Table3), particularly fatty acids such as oleic acid and linoleic acid, which are consistent with the expected composition of non-polar plant extracts (6, 35). These findings align with previous reports indicating that the hexane fraction of M. oleifera leaves is enriched in hydrophobic compounds, including long-chain fatty acids and related derivatives (6).
Compound identification as presented in Table 3, was based on spectral matching with the NIST library (32) (≥ 90% similarity). While this approach is widely used in phytochemical studies, it remains inherently tentative in the absence of retention index (RI) confirmation or authentic standards, which are recommended for improved reliability in GC–MS-based identification (33, 34). In the present study, retention indices were not determined, representing a limitation in compound verification.
Additionally, the detection of fatty acid methyl esters (FAMEs) may reflect in situ transesterification, thermal degradation during analysis, or library matching ambiguity, particularly in the absence of an explicitly described derivatization step. Similar observations have been reported in GC–MS analyses of plant oils and extracts, where methyl esters may arise as analytical artifacts or misassignments (33).
| Rt Time (min) | Compound Name | Molecular Formula | CAS Number | Peak Area (%) | *Quality Match |
|---|---|---|---|---|---|
| 31.283 | 10-Octadecenoic acid, methyl ester | C19H36O2 | 13481-95-3 | 23.78 | 98 |
| 31.122 | 14-Octadecenoic acid, methyl ester | C19H36O2 | 56554-48-4 | 20.10 | 98 |
| 27.383 | Pentadecanoic acid, 14-methyl-, methyl ester | C16H32O2 | 5129-60-2 | 7.38 | 97 |
| 27.727 | Hexadecanoic acid, methyl ester | C17H34O2 | 112-39-0 | 4.99 | 97 |
| 30.974 | 9,12-Octadecadienoic acid (Z, Z)-, methyl ester (Linoleate) | C19H34O2 | 112-63-0 | 1.51 | 98 |
| 27.583 | Hexadecanoic acid, methyl ester (Methyl palmitate) | C17H34O2 | 112-39-0 | 0.72 | 91 |
| 12.643 | 1,4-Methanonaphthalene, 1,4-dihydro- | C11H12 | 4453-90-1 | 0.16 | 93 |
| Note: The “Quality Match” refers to the confidence level of identification using the NIST library. | |||||
Quantitative interpretation of GC–MS data was also limited, as the peak area (shown in Table 3 and correspondingly indicated in Figure 2) normalization and calibration against reference standards were not performed. Therefore, relative abundance data is being interpreted cautiously and not considered definitive.
Collectively, LC–MS and GC–MS analyses indicate that MOHx contains a mixture of lipophilic and minor polar constituents, with fatty acids forming a dominant component. This compositional profile is consistent with the use of non-polar solvents such as hexane, which preferentially extract hydrophobic metabolites (6).
From a pharmaceutical perspective, the predominance of fatty acids—particularly unsaturated species such as oleic and linoleic acids, has important implications for formulation stability. These compounds are known to be susceptible to oxidative degradation (lipid peroxidation), which may compromise product stability, efficacy, and safety during processing and storage (36).
However, no stability studies were conducted in the present work to evaluate oxidative degradation or compositional changes during granulation and encapsulation. This represents a key limitation, as oxidative instability is a well-documented concern in lipid-rich formulations (8, 28).
In addition, the lack of quantitative phytochemical standardization further limits reproducibility and quality control, which are critical considerations in the development of phytopharmaceutical products (12). Future studies should therefore incorporate quantitative LC–MS/GC–MS validation, the use of authentic reference standards, determination of retention indices (GC–MS) and, stability testing under controlled conditions
Acute Toxicity Studies
Acute toxicity evaluation of MOHx was conducted using Lorke’s method. In Phase I, no mortality was observed at doses of 10.100, and 1.000 mg/kg. In Phase II, animals administered 1.600 and 2.900 mg/kg survived without mortality, although mild behavioral signs such as restlessness and pinna erection were noted. At the highest tested dose of 5.000 mg/kg, mortality was observed, preceded by clinical signs including gasping, abdominal writhing, pupil dilation, fur erection, and restlessness. Based on these observations, the median lethal dose (LD₅₀) for orally administered MOHx was estimated to be 3.808 mg/kg body weight, calculated as the geometric mean of the highest non-lethal dose (2.900 mg/kg) and the lowest lethal dose (5.000 mg/kg), in accordance with Lorke’s method (19). According to commonly referenced toxicity classification scales, substances with LD₅₀ values between 500 and 5.000 mg/kg are generally considered to exhibit low to moderate acute toxicity (37). Within this context, MOHx may be regarded as having relatively low acute toxicity at lower doses; however, the occurrence of mortality and pronounced behavioral disturbances at higher doses indicates that toxicity is dose-dependent and caution is warranted at elevated exposure levels.
A summary of the acute toxicity findings is presented in Table 4.
| Sample | Phase | Dose (mg/kg) | Death/no. Rat | Survival % (24 h) | Average Time of Death (h) | Behavioural Signs |
|---|---|---|---|---|---|---|
| MOHx | I | 10 | 0/3 | 100 | No death | - |
| 100 | 0/3 | 100 | No death | - | ||
| 1000 | 0/3 | 100 | No death | Restlessness | ||
| MOHx | II | 1600 | 0/1 | 100 | No death | Restlessness |
| 2900 | 0/1 | 100 | No death | Restlessness, erection of pinna | ||
| 5000 | 1/1 | 0 | Approx. 20hs | Pupil dilation, abdominal writhing, fur erection, gasping | ||
| Note: MOHx = Hexane fraction of M. oleifera leaves; LD50 = 3808 mg/kg | ||||||
Compared with previously reported aqueous and ethanolic extracts of M. oleifera, which often demonstrate wider safety margins, the hexane fraction appears to exhibit relatively higher toxicity at elevated doses (4, 7). This difference may be attributed to the enrichment of lipophilic constituents in the hexane fraction, including fatty acids and related compounds identified through LC–MS and GC–MS analyses. While such compounds (e.g., linoleic acid derivatives and jasmonates) are known to possess biological activity, their potential contribution to toxicity at high concentrations remains speculative and was not directly evaluated in this study.
Despite providing preliminary insight into the acute toxicity profile of MOHx, several important limitations should be acknowledged. The study relied solely on mortality endpoints and gross behavioral observations, without incorporation of detailed toxicological evaluations such as biochemical, hematological, or histopathological assessments, which are essential for comprehensive safety characterization. More so, the use of a single animal per dose level in Phase II, although consistent with Lorke’s method, limits the statistical robustness and reproducibility of the findings. Furthermore, the observation period was restricted to 24 h, whereas extended monitoring (e.g., up to 14 days as recommended in OECD guidelines) is typically required to capture delayed toxic effects (37). Therefore, while the estimated LD₅₀ of 3, 808 mg/kg suggests that MOHx exhibits relatively low acute toxicity within the tested dose range, this finding should be interpreted as preliminary rather than definitive evidence of safety. Comprehensive toxicological evaluation, including sub-acute and chronic toxicity studies, biochemical profiling, and organ histopathology, is required to establish a more reliable safety profile for the extract.
In silico Studies of LCMS-identified Phytomolecules of MOHx
Drug-likeness of Ligands and Physicochemical Properties
The physicochemical properties and drug-likeness profiles of selected MOHx-derived compounds are presented in Table 5. Most compounds complied reasonably with Lipinski’s rule of five, suggesting potential suitability for oral bioavailability (21). Specifically, jasmonic acid, citreoisocoumarin, and 2-furoic acid exhibited acceptable molecular weights, hydrogen bonding capacity, and lipophilicity, supporting favorable absorption characteristics.
In contrast, linoleic acid and oleic acid exceeded the recommended number of rotatable bonds, which may negatively influence conformational stability and oral bioavailability. Additionally, 7, 8-dihydropteroic acid showed a relatively high topological polar surface area (TPSA > 140 Ų), which may limit membrane permeability. These deviations indicate that while some compounds may not fully satisfy classical drug-likeness criteria, they could still serve as lead scaffolds or structural precursors for further optimization, consistent with earlier reports on natural product-based drug discovery (20).
| S/n | *Compounds from MOHx | Molecular weight (Da) | Rotatable bonds (KJmol-1) | Hydrogen bond acceptors (KJmol-1) | Hydro-gen bond donors (KJmol-1) | TPSA (Å2) | Molar refractivity (cm) | logPo/w |
|---|---|---|---|---|---|---|---|---|
| 1 | 7,8-Dihydropteroic acid | 314.3 | 4 | 5 | 5 | 145.49 | 92.52 | -1.28 |
| 2 | Jasmonic acid | 210.27 | 5 | 3 | 1 | 54.37 | 59.18 | 1.68 |
| 3 | Citreoisocourmarin | 146.14 | 0 | 2 | 0 | 30.21 | 42.48 | 1.65 |
| 4 | 2-furoic acid | 112.08 | 1 | 3 | 1 | 50.44 | 25.67 | -0.36 |
| 5 | Linoleic acid | 280.45 | 14 | 2 | 1 | 37.3 | 89.46 | 4.57 |
| 6 | Oleic acid | 282.46 | 15 | 2 | 1 | 37.3 | 89.94 | 4.57 |
| · Refers: Table 2; TPSA = Topological Polar Surfance Area | ||||||||
In silico Pharmacokinetic Profile
The predicted pharmacokinetic parameters (Table 6) indicate that most compounds, particularly jasmonic acid, citreoisocoumarin, 2-furoic acid, linoleic acid, and oleic acid, exhibit high gastrointestinal absorption, whereas 7,8-dihydropteroic acid showed comparatively low absorption. None of the compounds were predicted to be substrates of P-glycoprotein (P-gp), suggesting a reduced likelihood of active efflux and potential improvement in intracellular retention (20, 38). Regarding metabolic interactions, selective inhibition of cytochrome P450 isoforms (e.g., CYP1A2 and CYP2C9) was observed for some compounds (Table 6), indicating a possible risk of drug–drug interactions, as previously documented for phytochemicals affecting metabolic enzymes (39). Skin permeability values (log Kp) were uniformly low, suggesting limited suitability for transdermal delivery, but this parameter is of limited relevance given the intended oral dosage form. Overall, these pharmacokinetic predictions suggest moderate oral bioavailability potential for several compounds; however, such in silico estimates are inherently model-dependent and may not accurately reflect in vivo pharmacokinetics, particularly in complex plant extracts (39, 33).
| S/n | Compound | HIA | BBB | P-gp | Logkp | F | CYP1A2 | CYP2C19 | CYP2C9 | CYP2D6 | CYP3A4 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 7,8-Dihydropteroic acid | Low | No | No | -8.29 | 0.56 | No | No | No | No | No |
| 2 | Jasmonic acid | High | Yes | No | -6.45 | 0.85 | No | No | No | No | No |
| 3 | Citreoisocourmarin | High | Yes | No | -5.84 | 0.55 | Yes | No | No | No | No |
| 4 | 2-furoic acid | High | Yes | No | -6.53 | 0.85 | No | No | No | No | No |
| 5 | Linoleic acid | High | No | No | -2.6 | 0.85 | Yes | No | Yes | No | No |
| 6 | Oleic acid | High | No | No | -2.6 | 0.85 | Yes | No | Yes | No | No |
Molecular Docking Analysis
The molecular docking results (Table 7) demonstrated varying binding affinities of the selected compounds toward the NDV target protein. Among the compounds, 7, 8-dihydropteroic acid exhibited the strongest binding affinity (–8.4 kcal/mol), followed by citreoisocoumarin (–6.4 kcal/mol) and jasmonic acid (–6.1 kcal/mol), while fatty acids such as linoleic and oleic acids showed comparatively weaker interactions. Although binding energies below –7 kcal/mol are often considered indicative of favorable ligand–protein interactions (40), such thresholds should be interpreted cautiously. Docking scores are theoretical approximations and do not directly correspond to biological activity or antiviral efficacy (10). Furthermore, the suggestion that 7,8-dihydropteroic acid may interfere with viral processes, while plausible given its known role in microbial folate biosynthesis inhibition (40), remains speculative in the absence of experimental validation. Compounds with moderate to low binding affinities (as seen in Table 7), including fatty acids, may still contribute indirectly through non-specific mechanisms such as membrane interaction or modulation of host responses. However, lipophilic molecules are also known to exhibit non-specific binding behavior, which may overestimate their apparent docking performance (41). Importantly, these docking results are based on tentatively identified compounds (Table 2) and an unvalidated docking protocol, which further limits the reliability of structural and interaction predictions. Therefore, the findings should be regarded as preliminary and hypothesis-generating, rather than confirmatory evidence of antiviral potential.
| Compound ID | Compounds | Binding affinity (Kcal/mol) |
|---|---|---|
| 135398662 | 7,8-Dihydropteroic acid | -8.4 |
| 5281166 | Jasmonic acid | -6.1 |
| 68108 | Citreoisocourmarin | -6.4 |
| 6919 | 2-furoic acid | -4.7 |
| 5280450 | Linoleic acid | -5.2 |
| 445639 | Oleic acid | -5.4 |
Toxicity Prediction
The predicted toxicity profiles of individual compounds (Tables 8–13) revealed considerable variability across endpoints, reflecting the structural diversity of the phytochemicals. Fatty acids such as oleic acid and linoleic acid (Tables 8 and 9) were generally predicted to be non-toxic across most endpoints (42), although both showed potential ecotoxicity signals. These findings are broadly consistent with their established biological roles and relatively high LD₅₀ values (43).
| Classification | Target | Prediction | Probability |
|---|---|---|---|
| Organ toxicity | Hepatotoxicity | Inactive | 0.55 |
| Organ toxicity | Neurotoxicity | Inactive | 0.91 |
| Organ toxicity | Nephrotoxicity | Inactive | 0.55 |
| Organ toxicity | Respiratory toxicity | Inactive | 0.84 |
| Organ toxicity | Cardiotoxicity | Inactive | 0.99 |
| Toxicity end points | Carcinogenicity | Inactive | 0.64 |
| Toxicity end points | Immunotoxicity | Inactive | 0.99 |
| Toxicity end points | Mutagenicity | Inactive | 1 |
| Toxicity end points | Cytotoxicity | Inactive | 0.71 |
| Toxicity end points | BBB-barrier | Active | 0.89 |
| Toxicity end points | Ecotoxicity | Active | 0.65 |
| Toxicity end points | Clinical toxicity | Inactive | 0.61 |
| Toxicity end points | Nutritional toxicity | Inactive | 0.89 |
| Classification Classification | Target | Prediction | Probability |
|---|---|---|---|
| Organ toxicity | Hepatotoxicity | Inactive | 0.55 |
| Organ toxicity | Neurotoxicity | Inactive | 0.91 |
| Organ toxicity | Nephrotoxicity | Inactive | 0.55 |
| Organ toxicity | Respiratory toxicity | Inactive | 0.84 |
| Organ toxicity | Cardiotoxicity | Inactive | 0.99 |
| Toxicity end points | Carcinogenicity | Inactive | 0.64 |
| Toxicity end points | Immunotoxicity | Inactive | 0.96 |
| Toxicity end points | Mutagenicity | Inactive | 1 |
| Toxicity end points | Cytotoxicity | Inactive | 0.71 |
| Toxicity end points | BBB-barrier | Active | 0.89 |
| Toxicity end points | Ecotoxicity | Active | 0.65 |
| Toxicity end points | Clinical toxicity | Inactive | 0.61 |
| Toxicity end points | Nutritional toxicity | Inactive | 0.89 |
In contrast, 2-furoic acid (Table 10) exhibited predicted nephrotoxicity and carcinogenicity, aligning with previous reports on the toxicological potential of furan derivatives via oxidative stress and reactive metabolite formation (44–46). Similarly, citreoisocoumarin (Table 11) showed multiple predicted toxicities, including hepatotoxicity and neurotoxicity, consistent with known effects of coumarin-related compounds (47, 48).
| Classification | Target | Prediction | Probability |
|---|---|---|---|
| Organ toxicity | Hepatotoxicity | Inactive | 0.67 |
| Organ toxicity | Neurotoxicity | Inactive | 0.85 |
| Organ toxicity | Nephrotoxicity | Active | 0.68 |
| Organ toxicity | Respiratory toxicity | Inactive | 0.55 |
| Organ toxicity | Cardiotoxicity | Inactive | 0.84 |
| Toxicity end points | Carcinogenicity | Active | 0.51 |
| Toxicity end points | Immunotoxicity | Inactive | 0.99 |
| Toxicity end points | Mutagenicity | Inactive | 0.87 |
| Toxicity end points | Cytotoxicity | Inactive | 0.72 |
| Toxicity end points | BBB-barrier | Active | 0.91 |
| Toxicity end points | Ecotoxicity | Inactive | 0.60 |
| Toxicity end points | Clinical toxicity | Inactive | 0.68 |
| Toxicity end points | Nutritional toxicity | Inactive | 0.82 |
| Classification | Target | Prediction | Probability |
|---|---|---|---|
| Organ toxicity | Hepatotoxicity | Active | 0.69 |
| Organ toxicity | Neurotoxicity | Active | 0.87 |
| Organ toxicity | Nephrotoxicity | Inactive | 0.90 |
| Organ toxicity | Respiratory toxicity | Active | 0.98 |
| Organ toxicity | Cardiotoxicity | Inactive | 0.77 |
| Toxicity end points | Carcinogenicity | Inactive | 0.62 |
| Toxicity end points | Immunotoxicity | Active | 0.96 |
| Toxicity end points | Mutagenicity | Inactive | 0.97 |
| Toxicity end points | Cytotoxicity | Inactive | 0.93 |
| Toxicity end points | BBB-barrier | Inactive | 1 |
| Toxicity end points | Ecotoxicity | Active | 0.73 |
| Toxicity end points | Clinical toxicity | Inactive | 0.56 |
| Toxicity end points | Nutritional toxicity | Inactive | 0.74 |
Jasmonic acid (Table 12) demonstrated relatively low predicted toxicity despite some indications of respiratory and clinical effects, which may be related to its biological role in plant signaling and immune modulation (49, 50).
| Classification | Target | Prediction | Probability |
|---|---|---|---|
| Organ toxicity | Hepatotoxicity | Inactive | 0.77 |
| Organ toxicity | Neurotoxicity | Inactive | 0.89 |
| Organ toxicity | Nephrotoxicity | Inactive | 0.53 |
| Organ toxicity | Respiratory toxicity | Active | 0.54 |
| Organ toxicity | Cardiotoxicity | Inactive | 0.85 |
| Toxicity end points | Carcinogenicity | Inactive | 0.74 |
| Toxicity end points | Immunotoxicity | Inactive | 0.81 |
| Toxicity end points | Mutagenicity | Inactive | 0.88 |
| Toxicity end points | Cytotoxicity | Inactive | 0.66 |
| Toxicity end points | BBB-barrier | Active | 0.77 |
| Toxicity end points | Ecotoxicity | Inactive | 0.70 |
| Toxicity end points | Clinical toxicity | Active | 0.53 |
| Toxicity end points | Nutritional toxicity | Inactive | 0.78 |
Conversely, 7, 8-dihydropteroic acid (Table 13) exhibited multiple predicted toxicities, including hepatotoxicity and immunotoxicity, potentially linked to its interference with folate-related metabolic pathways (51).
| Classification Classification | Target | Prediction | Probability |
|---|---|---|---|
| Organ toxicity | Hepatotoxicity | Active | 0.69 |
| Organ toxicity | Neurotoxicity | Active | 0.87 |
| Organ toxicity | Nephrotoxicity | Inactive | 0.90 |
| Organ toxicity | Respiratory toxicity | Active | 0.98 |
| Organ toxicity | Cardiotoxicity | Inactive | 0.77 |
| Toxicity end points | Carcinogenicity | Inactive | 0.62 |
| Toxicity end points | Immunotoxicity | Active | 0.96 |
| Toxicity end points | Mutagenicity | Inactive | 0.97 |
| Toxicity end points | Cytotoxicity | Inactive | 0.93 |
| Toxicity end points | BBB-barrier | Inactive | 1 |
| Toxicity end points | Ecotoxicity | Active | 0.73 |
| Toxicity end points | Clinical toxicity | Inactive | 0.56 |
| Toxicity end points | Nutritional toxicity | Inactive | 0.74 |
While these predictions provide useful preliminary insights, it is important to recognize that in silico toxicity models are optimized for individual compounds and may not accurately represent the safety profile of complex mixtures such as MOHx (11). This limitation is further highlighted by the disparity between predicted LD₅₀ values and the experimentally determined extract-level LD₅₀ reported in this study (Table 4).
Formulation Study of MOHx
pH of the Extract
The pH of MOHx was determined to be 6.98, indicating a near-neutral character. This range is generally considered acceptable for oral formulations, as it reduces the likelihood of gastrointestinal irritation and supports compatibility with a wide range of excipients used in capsule formulations. From a stability perspective, near-neutral pH conditions are less likely to promote hydrolytic degradation compared to highly acidic or alkaline environments (26). However, it should be noted that pH alone does not fully predict chemical stability, particularly for lipophilic constituents that may undergo oxidative degradation independent of aqueous conditions.
Solubility Test
The solubility profile of MOHx (Table 14) confirmed its poor aqueous solubility, as the extract remained insoluble across aqueous media, including acidic (0.1 N HCl), alkaline (0.1 N NaOH), buffered (pH 4.0, 6.8, 8.0), and isotonic (0.9% NaCl) environments, while showing solubility only in ethanol. This behavior is consistent with the predominance of lipophilic constituents identified in the extract (Table 2), such as fatty acids and related non-polar compounds. Poor aqueous solubility is a well-recognized limitation for oral delivery, as it may restrict dissolution in gastrointestinal fluids and consequently reduce bioavailability (27). The findings therefore justify the selection of formulation strategies aimed at improving dispersion and dissolution behavior, rather than implying intrinsic solubility enhancement. Similar solubility challenges have been widely reported for non-polar plant fractions, necessitating the use of granulation and hydrophilic excipients to facilitate drug release (26, 27).
| Test | Observation | Inference |
|---|---|---|
| MOHx + 0.1N HCl | No solubility | Insoluble |
| MOHx + 0.1N NaOH | No solubility | Insoluble |
| MOHx + Buffer (pH 4.0, 6.8, 8.0) | No solubility in all | Insoluble |
| MOHx + 99.7% Ethanol | Solubility formed in excess solvent | Soluble |
| MOHx + 0.9% NaCl | No solubility | Insoluble |
Micrometric Properties of the Granules of MOHx
The micromeritic properties of MOHx granules across all batches are presented in Table 15. Bulk density (0.31–0.46 g/cm³) and tapped density (0.41–0.54 g/cm³) fell within pharmacopeial ranges for powder flow systems, indicating acceptable packing characteristics (28-30).
Flowability indices, including Carr’s compressibility index (14.82–25.30%) and Hausner’s ratio (1.17–1.34), suggest fair to good flow properties. While values below 25% (Carr’s index) and 1.25 (Hausner’s ratio) are typically indicative of good flow, values approaching the upper limits (as seen in Batch IV) suggest only passable flow, which may require careful handling during large-scale processing (29, 30). Similarly, the angle of repose values (36.2°–39.5°) indicate moderate flowability, rather than excellent flow.
These findings demonstrate that wet granulation improved powder handling properties sufficiently for capsule filling, consistent with previous reports on herbal formulations (13, 14). However, the results also highlight that flow properties were not optimal, particularly in higher-density batches, suggesting that further optimization (e.g., glidant concentration adjustment) may be beneficial for scale-up.
| Parameter | Batch I | Batch II | Batch III | Batch IV | Reference Value | Inference |
|---|---|---|---|---|---|---|
| Bulk Density (g/dm3) | 0.46 | 0.395 | 0.315 | 0.310 | 0.2-0.8 | Passed |
| Tapped Density (g/dm3) | 0.54 | 0.495 | 0.410 | 0.415 | 0.4-1.0 | Passed |
| Hausner’s ratio | 1.17 | 1.25 | 1.3 | 1.34 | 1.1-1.5 | Passed |
| C.C.I (Carr’s compressibility index (%) | 14.82 | 20.82 | 23.17 | 25.30 | 5-25 | Passed |
| Angle of Repose (°) | 36.2 | 38.1 | 37.6 | 39.5 | 25-40 | Passed |
Weight Variation
The weight variation results (Table 16) showed that all capsule batches complied with pharmacopeial specifications, with percentage deviations ranging from 4.5% to 7.0%, well within the ±10% limit for hard gelatin capsules (26). This level of uniformity reflects adequate flow and die-filling consistency, which are critical for ensuring dose reproducibility, particularly in herbal formulations with inherently variable bulk densities (12). While the results indicate good process control at laboratory scale, it should be noted that content uniformity was not directly measured, and therefore uniform distribution of active constituents within capsules cannot be conclusively established.
| Batch | Average Weight (mg) | % Weight Variation | Reference Value | Inference |
|---|---|---|---|---|
| I | 205 | 4.5 | < 10% | Passed |
| II | 200 | 5.0 | < 10% | Passed |
| III | 210 | 7.0 | < 10% | Passed |
| IV | 207 | 5.2 | < 10% | Passed |
Disintegration Time
Disintegration times for all batches (Table 17) ranged from 27.40 to 29.62 min, complying with the pharmacopeial limit of less than 30 min for hard gelatin capsules (27). However, these values are at the upper threshold of acceptability, indicating relatively slow disintegration. The prolonged disintegration, particularly in Batch IV, may be attributed to higher concentrations of excipients such as microcrystalline cellulose, which can enhance mechanical strength but delay water penetration and capsule rupture (14). Comparable disintegration profiles have been reported for herbal capsule formulations containing poorly soluble extracts (11). Although the results meet regulatory requirements, faster disintegration is generally desirable to promote timely drug release. Future formulation optimization could incorporate superdisintegrants (e.g., croscarmellose sodium) to improve disintegration efficiency without compromising granule integrity.
| Batch | Average Disintegration Time (Min) | Reference Value (Min) | Inference |
|---|---|---|---|
| I | 27.40 | < 30 | Passed |
| II | 28.70 | < 30 | Passed |
| III | 28.92 | < 30 | Passed |
| IV | 29.62 | < 30 | Passed |
Dissolution Profile
The dissolution profile (Figure 3) demonstrated that more than 80% of the extract was released within 20 min, exceeding the pharmacopeial requirement of at least 70% release within 45 min for immediate-release formulations (26, 27). This indicates that the formulation approach—particularly the use of wet granulation and hydrophilic excipients—successfully enhanced drug dispersion and release behavior. Such improvements are consistent with previous studies showing that granulation techniques can significantly improve the dissolution of poorly water-soluble phytoconstituents by increasing wettability and surface area (13, 14). In contrast, direct compression methods have been associated with slower and less reproducible dissolution profiles for lipophilic herbal extracts (29). However, it is important to emphasize that enhanced dissolution does not necessarily translate to improved in vivo bioavailability, particularly for lipophilic compounds whose absorption may be limited by permeability and first-pass metabolism (14). Additionally, the study did not include comparative dissolution testing with the unformulated extract, which limits the ability to quantify the extent of improvement achieved.
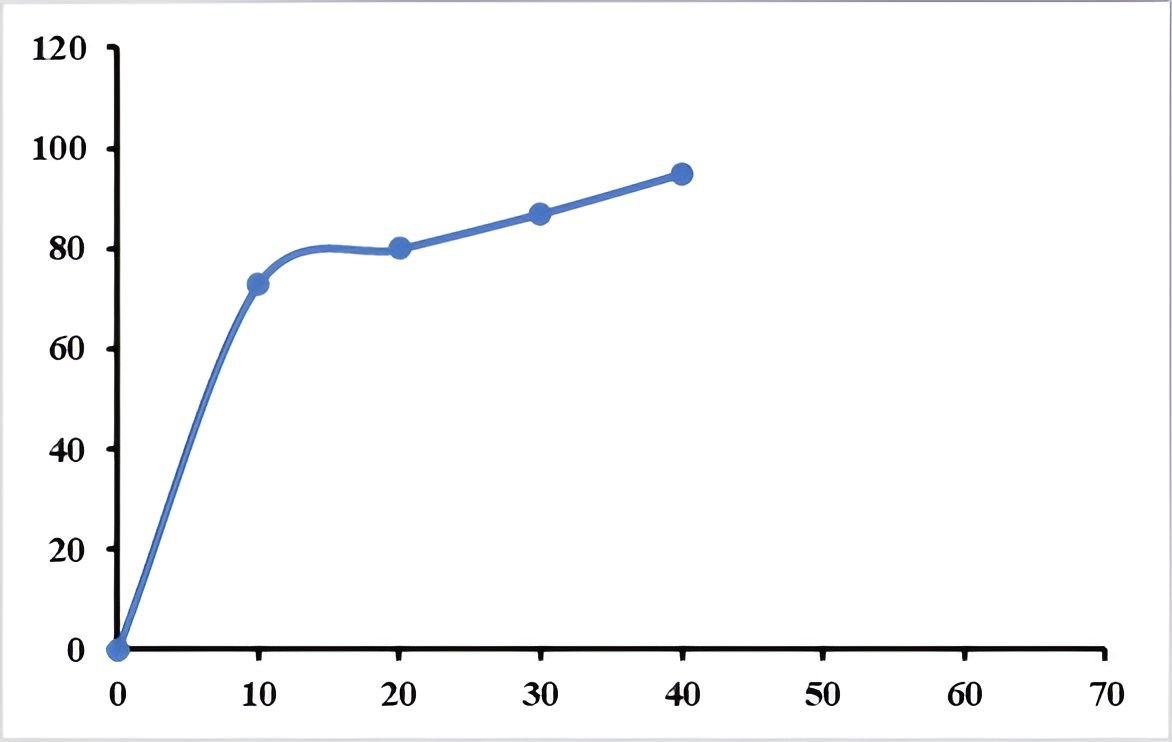
The overall formulation results (Tables 14–17; Figure 3) indicate that the developed MOHx capsules comply with key pharmacopoeial quality requirements, including acceptable flow properties, uniformity of weight, appropriate disintegration time, and satisfactory dissolution performance. These findings suggest that the application of wet granulation was effective in improving the handling characteristics of the extract and enhancing its release profile, particularly in light of the inherent challenges associated with lipophilic plant-derived materials. However, despite these favorable outcomes, certain limitations of the study should be recognized. Notably, quantitative data demonstrating the extent of solubility enhancement were not established, and content uniformity testing was not performed, thereby limiting conclusions regarding dose consistency of active constituents. In addition, the absence of stability studies precludes assessment of the long-term physicochemical integrity of the formulation. Furthermore, while the in vitro dissolution profile was satisfactory, its direct correlation with in vivo bioavailability remains uncertain, as dissolution alone does not fully predict absorption behaviour, particularly for poorly water-soluble compounds (14). In view of these considerations, although the formulation strategy appears promising in improving the pharmaceutical handling and release characteristics of MOHx, further investigations are necessary to comprehensively establish its bioavailability, stability, and reproducibility under practical storage and physiological conditions.
Conclusion
This study demonstrated that the hexane fraction of M. oleifera leaves (MOHx) contains predominantly lipophilic phytoconstituents and can be formulated into a solid oral dosage form with acceptable pharmacopoeial quality attributes. The capsules produced via wet granulation exhibited satisfactory flowability, weight uniformity, disintegration, and dissolution performance (Tables 14–17; Figure 3), indicating that the formulation strategy effectively addressed key handling and release challenges associated with poorly water-soluble plant extracts.
These findings support the study’s objective of developing a standardized and pharmaceutically acceptable herbal capsule from MOHx, while also underscoring the importance of formulation techniques in enhancing the performance of lipophilic phytocompounds. Within the broader context of herbal drug development, this work contributes to ongoing efforts to improve the quality, reproducibility, and dosage form acceptability of plant-based therapeutics.
Nonetheless, the absence of stability data, content uniformity testing, and in vivo validation highlights important limitations that constrain the full assessment of the formulation’s therapeutic potential. Future studies should therefore focus on pharmacokinetic evaluation, long-term stability, and advanced formulation approaches to further optimize bioavailability and ensure product consistency.
Overall, this study establishes a strong and reliable foundational framework for the rational development of MOHx capsules and further reinforces the critical and essential role of pharmaceutical standardization in successfully translating bioactive plant extracts into clinically relevant and therapeutically effective dosage forms.
Abbreviation
ADME = Absorption, Distribution, Metabolism, and Excretion; BBB = Blood–Brain Barrier; BP = British Pharmacopoeia; CA = Carr’s Index; CCI = Carr’s Compressibility Index; CYP = Cytochrome P450; ESI = Electrospray Ionization; GC-MS = Gas Chromatography–Mass Spectrometry; HIA = Human Intestinal Absorption; HPLC = High-Performance Liquid Chromatography; LC-MS = Liquid Chromatography–Mass Spectrometry; LD₅₀ = Median Lethal Dose; log Kp = Skin Permeability Coefficient; log P (Po/w) = Octanol–Water Partition Coefficient; MOHx (MOHX) = n-Hexane Fraction of Moringa oleifera Leaves; MS/MS = Tandem Mass Spectrometry; NDV = Newcastle Disease Virus; NIST = National Institute of Standards and Technology; P-gp = P-glycoprotein; PDA = Photodiode Array Detector; Rt = Retention Time; TIC = Total Ion Chromatogram; TPSA = Total Polar Surface Area; UV–Vis = Ultraviolet–Visible Spectrophotometry.
Declarations
Acknowledgment
We appreciate the support by the management of Usmanu Danfodiyo University, Sokoto (UDUS), for facilitating the release of funds from Tetfund, as well as the technical assistance rendered by laboratory staff of UDUTH
Conflict of Interest
The authors declare no conflicting interest.
Data Availability
The authors confirm that the data supporting the findings of this study are available within the article.
Ethics Statement
This study received a prior-full consent approval from the Health Research Ethics Committee (HREC) of the Usmanu Danfodiyo University, Sokoto-Nigeria, with reference number: UDUS/UREC/2020/006 dated 20/10/2020
Funding Information
This work was supported by the [Tertiary Education Trust Fund (Tetfund)] under the National Research Fund (NRF) Grant Number [TETF/ES/DR&D-CE/NRF2020/SETI/110/VOL.1].
Supplemental Material
This manuscript contains two items of supplementary data: (1) <a class="cursor-pointer" href="https://etflin.com/file/document/20260328154624_76690_6c234635.docx">LCMS precursor and product ions</a>, and (2) the <a class="cursor-pointer" href="https://etflin.com/file/document/20260328154728_507694_16d187cf.docx">calibration curve of MOHx</a>.
References
- Akunna GG, Lucyann CA, Saalu LC. Rooted in tradition, thriving in the present: the future and sustainability of herbal medicine in Nigeria’s healthcare landscape. J Innov Med Res. 2023;2(11):28–40.
- El Bilali H, Guimbo ID, Nanema RK, Falalou H, Kiebre Z, Rokka VM, et al. Research on Moringa oleifera Lam. in Africa. Plants. 2024;13:1613.
- Mwami B, Maňourová A, Hendre PS, Muchugi A, Verner V, Kariuki P, et al. Traditional knowledge, use, and management of Moringa oleifera among the Mijikenda community in Kilifi, Kenya. Plants. 2024;13:3547.
- Maizuwo AI, Hassan AS, Momoh H, Muhammad JA. Phytochemical constituents, biological activities, therapeutic potentials and nutritional values of Moringa oleifera (Zogale): a review. J Drug Des Med Chem. 2017;3:60–66.
- Ofesi AO, Igere BE, Onoriasakpobare FO, Chukwuka EG. Antimicrobial and phytochemical analysis of Moringa oleifera seeds collected at the University of Ilorin Botanical Center, Kwara State, Nigeria. FUDMA J Sci. 2025;9:167–171.
- Arguelles-Peña K, Olguín-Rojas JA, Acosta-Osorio AA, Carrera C, Barbero GF, García-Alvarado MA, et al. Evaluation of equilibrium properties in hexane and ethanol extractive systems for Moringa oleifera seeds and fatty acid profiles of the extracts. Separations. 2021;8(11):217.
- Adebayo IA, Arsad H, Kamal N, Samian MR. The hexane fraction of Moringa oleifera Lam. seed extract induces apoptosis, causes cell cycle arrest, and modulates expression of HSP60, NPM, PGK1, RCN1, and PDIA1 in MCF7 cells. S Afr J Bot. 2020;129:379–387.
- Fahmy NM, Fayez S, Mohamed RW, Elissawy AM, Eldahshan OA, Zengin G, et al. Moringa oleifera flowers: insights into aroma chemistry, anti-inflammatory, antioxidant and enzyme inhibitory properties. BMC Complement Med Ther. 2024;24:286.
- Naik GG, Sahu AN, Kaushik V, Kaushik A, Sarkar BK. Phytopharmaceuticals and herbal drugs: prospects and safety issues in the delivery of natural products. In: Phytopharmaceuticals and herbal drugs. 2023. p. 215–245.
- Wang H, Chen Y, Wang L, Liu Q, Yang S, Wang C. Advancing herbal medicine: enhancing product quality and safety through robust quality control practices. Front Pharmacol. 2023;14:1265178.
- Jyothi D, Koland M, Priya S, James JP. Formulation of herbal capsules containing Trigonella foenum-graecum seed extract for the treatment of diabetes. J Young Pharm. 2017;9:352–356.
- Archer MA, Kumadoh D, Yeboah GN, Kyene MO, Kumatia EK, Antwi S, et al. Formulation and evaluation of capsules containing extracts of Cassia sieberiana for improved therapeutic outcome. Sci Afr. 2020;10:e00609.
- Osei-Asare C, Owusu FWA, Entsie P, Annan AK, Gyamaa RA, Amenuke EM. Formulation and in vitro evaluation of oral capsules from liquid herbal antimalarials marketed in Ghana. J Trop Med. 2021;2021:6694664.
- Van der Merwe J, Steenekamp J, Steyn D, Hamman J. The role of functional excipients in solid oral dosage forms to overcome poor drug dissolution and bioavailability. Pharmaceutics. 2020;12:393.
- Harborne JB. Phytochemical methods: a guide to modern techniques of plant analysis. London: Chapman & Hall; 1998.
- Mathias SN, Ilyas N, Musa KY. Chemical Society of Nigeria. 2007;4(1):70–75.
- Shah B, Seth A. Textbook of pharmacognosy and phytochemistry. Elsevier; 2010.
- Garba S, Adamu M, Sani MS, Muhammad MA. Advances in LC-MS analysis for phytochemical profiling of medicinal plants. J Anal Sci Technol. 2023;14:56.
- Lorke D. A new approach to practical acute toxicity testing. Arch Toxicol. 1983;54(4):275–287.
- Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717.
- Lipinski CA. Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov Today Technol. 2000;1:337–341.
- Gustafson CL, Partch CL, Crane BR. Structural biology of the protein data bank and molecular docking. Nat Struct Mol Biol. 2014;21(1):25–30.
- Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30:2785–2791.
- Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function. J Comput Chem. 2010;31:455–461.
- Priyanka N, Baburao P, Kumar JM, Priya KP. Protox-II: a web server for the prediction of toxicity of chemicals. J Comput Biol. 2018;25:524–533.
- British Pharmacopoeia Commission. British Pharmacopoeia. London: TSO; 2023.
- United States Pharmacopeial Convention. United States Pharmacopeia and National Formulary (USP 46–NF 41). Rockville (MD): United States Pharmacopeial Convention; 2023.
- Aulton ME, Taylor KMG. Aulton’s pharmaceutics: the design and manufacture of medicines. 5th ed. London: Elsevier; 2018.
- Banker GS, Anderson NR. Tablets. In: Lachman L, Lieberman HA, Kanig JL, editors. The theory and practice of industrial pharmacy. 4th ed. New York: Marcel Dekker; 2013. p. 293–345.
- Alderborn G. Granulation and tablet characteristics. Int J Pharm. 2013;456(1–2):227–236.
- Kind T, Fiehn O. Metabolomic database annotations via mass spectral matching. BMC Bioinformatics. 2007;8:105.
- Stein SE. NIST mass spectral database and software. Gaithersburg: National Institute of Standards and Technology; 2017.
- Wolfender JL, Nuzillard JM, van der Hooft JJJ, Renault JH, Bertrand S. Accelerating metabolite identification in natural product research. Anal Chem. 2019;91(1):704–742.
- Niessen WMA. Liquid chromatography–mass spectrometry. 3rd ed. Boca Raton: CRC Press; 2016.
- Christie WW. Lipid analysis: isolation, separation, identification. 3rd ed. Oily Press; 2003.
- Choe E, Min DB. Mechanisms of lipid oxidation. Compr Rev Food Sci. 2006;5:169–186.
- OECD. Acute oral toxicity guideline 423. OECD; 2001.
- Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2(1):48–58.
- Ekins S, Stresser DM, Williams JA. In vitro and pharmacophore insights into cytochrome P450-mediated drug–drug interactions. Br J Pharmacol. 2007;152(1):9–20.
- Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery. Nat Rev Drug Discov. 2004;3:935–949.
- Lounkine E, Keiser MJ, Whitebread S, Mikhailov D, Hamon J, Jenkins JL, et al. Large-scale prediction and testing of drug activity on side-effect targets. Nature. 2012;486:361–367.
- Lima TM, Kanunfre CC, Pompéia C, Verlengia R, Curi R. Ranking the toxicity of fatty acids on Jurkat and Raji cells by flow cytometric analysis. Toxicol In Vitro. 2015;16(6):741–747.
- Bergqvist J, Lydy MJ, de Perre C. Toxicity of fatty acids to aquatic organisms: a review. Environ Toxicol Chem. 2020;39(8):1482–1493.
- Navarro A, Tort-Navas FJ. Furan and renal damage: toxicological mechanisms. Toxicol Rep. 2010;7(1):12–19.
- Chen LJ, Zhang X, Zhang B, Wang L. Furan derivatives: toxicology, occurrence and chemistry. Compr Rev Food Sci. 2014;13(6):974–989.
- Peterson LA, Cummings ME, Vu CC, Matter BA. Glutathione trapping to investigate the role of reactive metabolites in drug-induced hepatotoxicity. Chem Res Toxicol. 2011;24(12):1970–1980.
- Duarte S, Kwee I, Wyman AR, Tavares W. Neurotoxicity of coumarins: mechanisms and prevention. Toxicol Lett. 2015;238(2):95–105.
- Lake BG. Coumarin metabolism, toxicity and carcinogenicity: relevance for human risk assessment. Food Chem Toxicol. 2009;47(4):691–702.
- Wasternack C, Hause B. Jasmonates: biosynthesis, perception, signal transduction and action in plant stress response, growth and development. Ann Bot. 2013;111(6):1021–1058.
- Gomes B, Figueiredo C, Simões S. Plant-derived molecules as modulators of inflammation and immunity. Phytochem Rev. 2019;18(5):1295–1317.
- Beardsley GP, Moroson BA, Taylor EC, Moran RG. Mechanism of action of folate antagonists. In: Antifolate drugs in cancer therapy. 2013. p. 11–44.