RESEARCH ARTICLE
Computational Assessment of Plant-Derived Alkaloids for Anti-SARS-CoV-2 Properties
Sciences of Phytochemistry|Vol. 5, Issue 2, pp. 249-261 (2026)
CC BY 4.0-2026 Authors
Views
Downloads
Shares
Received
Mar 11, 2026Revised
May 18, 2026Accepted
Jun 10, 2026Published
Jul 12, 2026
Abstract
The demand for efficient antiviral treatments that go beyond traditional treatment, whose effectiveness may be compromised by viral changes and documented adverse effects following immunization, has increased due to the quick and ongoing introduction of SARS-CoV-2 variations. In this regard, a potential class of natural chemicals for SARS-CoV-2 medication development is plant-derived alkaloids, which are known for their broad-spectrum antiviral qualities. This research employs network pharmacology, gene ontology, and molecular docking to find effective alkaloid candidates that can target conserved viral proteins across SARS-CoV-2 variations. 5 bioactive alkaloids were screened against three key coronavirus proteins—2AJF (spike RBD–ACE2 complex), 2DD8 (spike RBD-neutralizing antibody complex), and 2J98 (replication-associated nsp9 protein). Binding affinities ranged from -6.0 to -11.3 kcal/mol, with Manzamine A emerging as the strongest inhibitor across all targets (2AJF: -11.3, 2DD8: -10.0, 2J98: -9.1 kcal/mol). Its stable hydrogen-bonding network and interactions with key amino acids suggest strong potential to disrupt viral entry, immune evasion mechanisms, and replication processes. Network pharmacology revealed 5, 272 SARS-CoV-2 associated genes, of which 51 overlapped with alkaloid-related targets. Venn analysis identified 6 shared genes, and protein-protein interaction (PPI) network construction highlighted critical hub regulators like MTOR, AKT1, STAT3, SYK, CASP3, and JAK2 implicated in immune modulation, apoptosis, inflammation, and viral pathogenicity. The combined computational approach identifies Manzamine A as a promising natural scaffold for anti-SARS-CoV-2 drug development. Experimental validation through in vitro, in vivo, MD simulation and pharmacokinetic research is still necessary to show clinical application, even though the results provide insightful information for future therapeutic design.
Keywords:
Introduction
Since January 2020, the coronavirus SARS‑CoV-2 has caused COVID‑19 disease. New viral variants continue to arise through genetic changes. As of September 2025, XFG and NB.1.8.1 were the most common lineages worldwide, but current assessments show they do not have increased transmissibility, immune evasion, or severity compared with other circulating variants (1). The treatments have caused some adverse effects, especially those with pre-existing comorbidities (2, 3). There remains a risk of continued SARS‑CoV transmission, and viral mutations could produce new variants (4). Therefore, it is essential to explore alternative or complementary therapeutic approaches to control viral dissemination. Plant-derived alkaloids have been reported to possess significant antiviral properties, indicating their potential as bioactive compounds for therapeutic intervention (5, 6). Alkaloids that demonstrate high inhibitory activity against the SARS-CoV-2 virus can serve as potential candidates for managing future waves of coronavirus infection. Notably, viral protein structures such as 2AJF, 2DD8, and 2J98, which are associated with the SARS-CoV-2 spike and main protease regions, have conserved domains across multiple variants (7, 8).
Therefore, alkaloid compounds exhibiting strong binding affinities and stable interactions with these conserved proteins may be valuable in developing effective antiviral therapeutics (9). In order to determine which compounds had the greatest and lowest binding affinities, network pharmacology and molecular docking were used to examine the binding energies between certain alkaloids and SARS-CoV-2 viral proteins. The findings from such computational analyses can provide crucial insights for the rational design of plant-based antiviral drugs targeting SARS-CoV-2 and its emerging variants (10).
Methodology
Network Pharmacology
Employing Homo sapiens as the chosen organism, the Swiss Target Prediction platform (https://www.swisstargetprediction.ch/) was used to find possible protein targets using the SMILES structures of the chosen alkaloids that were taken from the PubChem database. In parallel, only entries classified as protein-coding genes were taken into consideration when retrieving disease-associated genes from the GeneCards database (https://www.genecards.org). The Jvenn (https://jvenn.toulouse.inrae.fr/app/example.html) diagram tool was used to display the crossing targets of each corona disease and the chosen alkaloids. The STRING database (https://string-db.org/) was used to build a protein–protein interaction (PPI) network utilizing the common target proteins and a high-confidence interaction score criterion (>0.9). Cytoscape (version 3.10.4) was used to visualize the final network. Using Cytoscape's String app plugin, topological analysis was carried out to find important regulatory genes. Network nodes were ranked according to radiality centrality, degree, and proximity metrics. Hub genes were defined as genes that consistently scored highest across all three categories. These genes were then linked with the molecular docking studies (6, 7).
Analysis of Gene Ontology and KEGG Pathway Enrichment
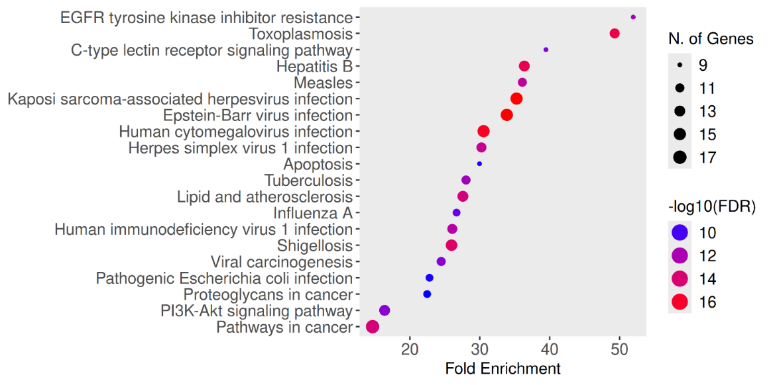
Using Gene Ontology (GO) and Kyoto Encyclopaedia of Genes and Genomes (KEGG) pathway frameworks, extensive functional enrichment analyses were carried out to clarify the biological importance of specific alkaloids predicted molecular interactions. ShinyGO v0.85 (https://bioinformatics.sdstate.edu/go/) and SRPLOT (https://www.bioinformatics.com.cn/en), two reputable bioinformatics platforms intended for high-throughput gene-set enrichment and route visualization, were used for the analyses. In order to identify probable biological functions and mechanistic correlations, enrichment analysis was performed on 52 intersecting genes that were found to be common targets between certain alkaloids and the corona illness. Within the GO framework, enriched terms were systematically categorized into the three principal ontological domains: Biological Process (BP), reflecting the physiological roles and pathways in which the genes are involved; Cellular Component (CC), denoting the subcellular localization or structural contexts of gene products; and Molecular Function (MF), representing the biochemical activities of these targets at the molecular level.
KEGG pathway analysis was subsequently utilized to delineate the most significantly enriched signalling and metabolic pathways potentially modulated by the selected alkaloids. A false discovery rate (FDR) threshold of less than 0.05 was used to evaluate statistical significance in order to guarantee strong control of multiple testing mistakes. The analytical results were visually represented through dot plots and bar charts, generated using ShinyGO and SRPLOT, providing an interpretable overview of the enriched functional categories and pathways associated with the identified target genes.
Molecular Docking
The Protein Data Bank provided the three-dimensional crystal structure of the unliganded hydrolase viral protein (2AJF), immune system viral protein (2DD8), and RNA binding protein (2J98) of the SARS-CoV-2 spike receptor binding domains for molecular docking studies (6, 11, 12). The human receptor ACE2 (angiotensin converting enzyme 2) forms a complex with the 2AJF, a SARS-CoV-2 spike protein receptor-binding domain. It is essential for developing vaccines and therapeutic antibodies as well as for comprehending how coronaviruses, such as SARS-Cov-2, attach to host cells. The crystal structure of the SARS-CoV-2 S protein receptor-binding domain (RBD) associated to the neutralizing antibody m396 is called 2DD8 (Crystal Structure of SARS-CoV-2 Spike Receptor-Binding Domain Complexed with Neutralizing Antibody). Researchers were able to comprehend the precise locations (epitopes) on the virus that the immune system targets in order to prevent infection thanks to this structure (13).
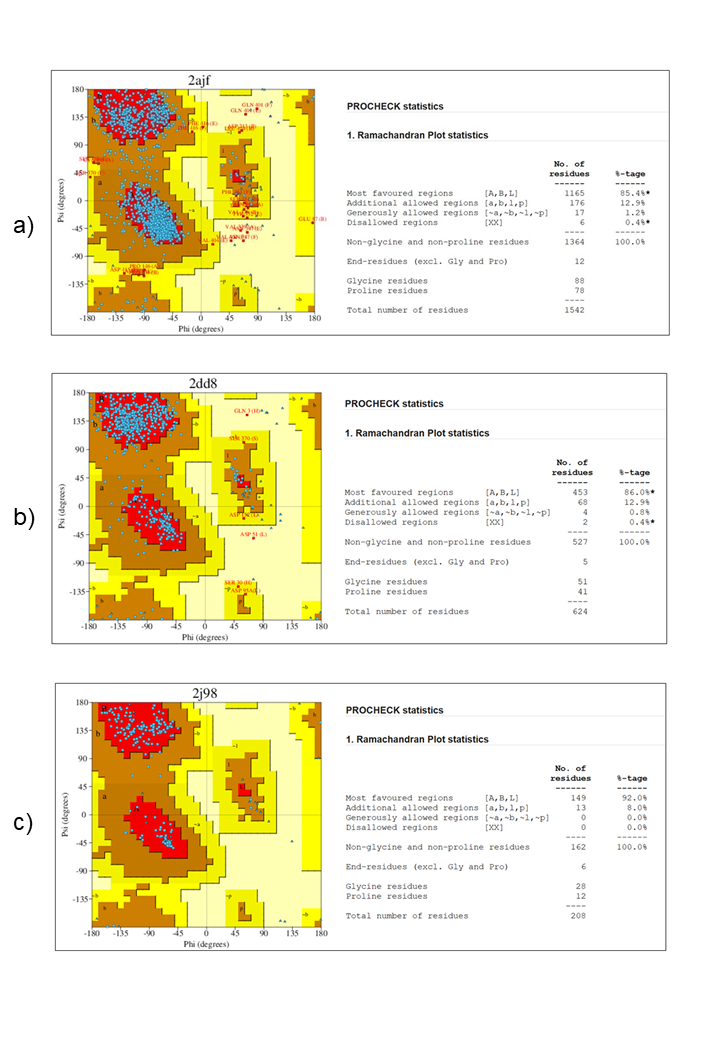
This information is critical for the design of antibody-based therapeutics and vaccines (14). 2J98 (Crystal structure of the 229E-CoV nsp9 C69A mutant) this non-structural protein 9 (nsp9) from the human coronavirus 229E (HCoV-229E), a different type of coronavirus. By binding to single-stranded RNA to form a homodimer a combination of two identical protein units the nsp9 protein plays a crucial part in viral replication (15). One possible aim for creating broad anti-coronavirus treatments is to interfere with this dimerization. All non-essential water molecules and heteroatoms were carefully removed using Molegro Molecular Viewer (MMV) to refine and prepare the target protein, which was then saved in PDB format. The active site characteristics, including the geometry and volume of the binding pocket, were identified using the CASTp server (16). The protein’s structural quality was assessed using the PROCHECK server (17).
Ramachandran plot analysis confirmed that more than 90% of the amino acid residues were located within energetically favorable regions. This distribution, along with the observation of minimal steric clashes in the psi (ψ) and phi (φ) torsion angles, indicated a high degree of stereochemical accuracy and overall structural stability of the protein (9). A collection of five antiviral alkaloid compounds from various plant sources (18), namely Anisomycin, Camptothecin, Fangchinoline, Manzamine A, and Tetrandrine, were acquired from the PubChem database (19). To ensure compatibility for docking techniques, Molegro Molecular Viewer was used to convert these compounds, which were initially available in Structure Data File (SDF) format, to Protein Data Bank (PDB) format. Molecular docking simulations were then conducted using PyRx 0.8, with ligand structures imported and optimized through Open Babel for accurate binding evaluation (20). A docking grid was generated, and molecular interactions were assessed through the Vina wizard. The docking outcomes were visualized and analyzed using Discovery Studio 2025, which provided an in-depth representation of 2D and 3D molecular interactions between 2AJF, 2DD8 and 2J98, with the ligands, offering insights into their binding affinities and structural compatibilities (21).
Results
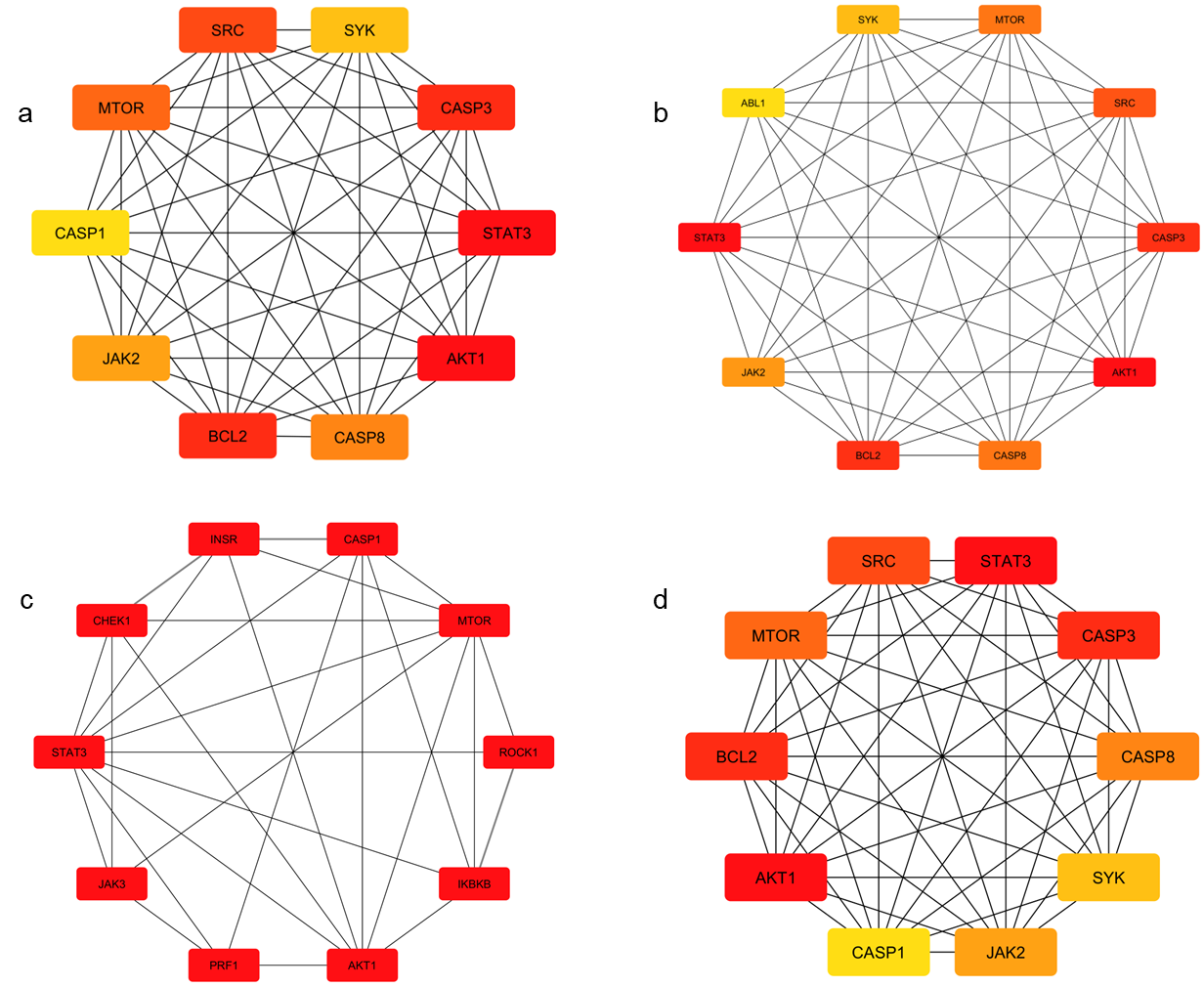
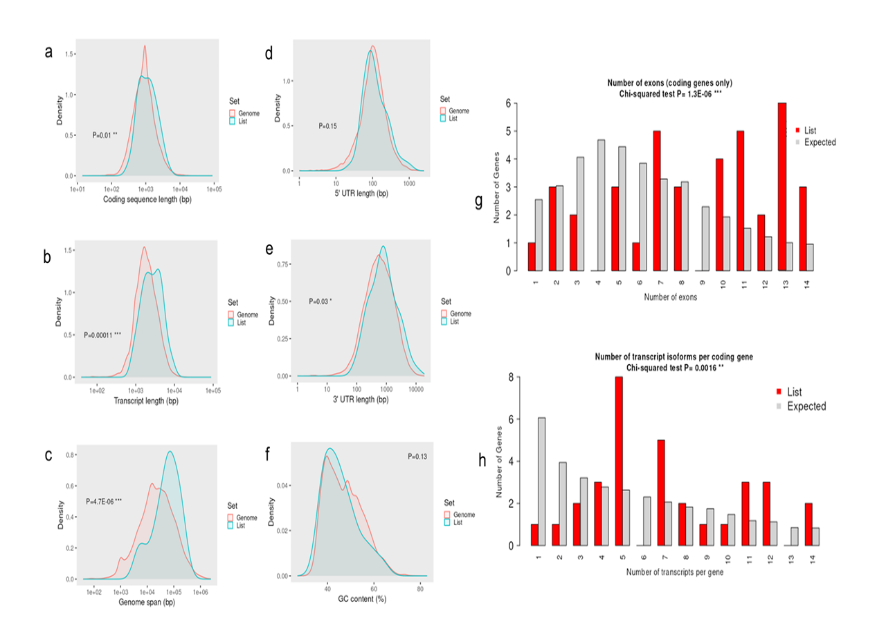
In the network pharmacology analysis, a total of 5, 272 disease-associated genes were identified for the SARS-CoV-2 variant. Among these, 51 genes were found to be related to the selected alkaloids. Venn diagram analysis revealed 6 overlapping genes shared between the alkaloid-associated genes and disease-related genes, indicating their potential as therapeutic targets (Table 1; Figures 1 and 2) (14). These intersecting targets showed a broad pattern of interaction when the Protein-Protein Interaction (PPI) network was constructed (Figure 3). Furthermore, network analysis emphasized the critical involvement of key signaling molecules such as MTOR, STAT3, AKT1, SYK, CASP3, and JAK2 (Figure 4), suggesting their possible roles in mediating the therapeutic effects of the selected alkaloids against the SARS-CoV-2.
| No. | Compound | Binding affinity (Kcal/mol) | ||
|---|---|---|---|---|
| 2AJF | 2DD8 | 2J98 | ||
| 1. | Reserpine | -8.0 | -8.6 | -7.8 |
| 2. | Camptothecin | -8.4 | -8.2 | -7.7 |
| 3. | Tetrandrine | -10.3 | -9.4 | -7.5 |
| 4. | Manzamine A | -11.3 | -10.0 | -9.1 |
| 5. | Fangchinoline | -10.5 | -9.3 | -7.6 |
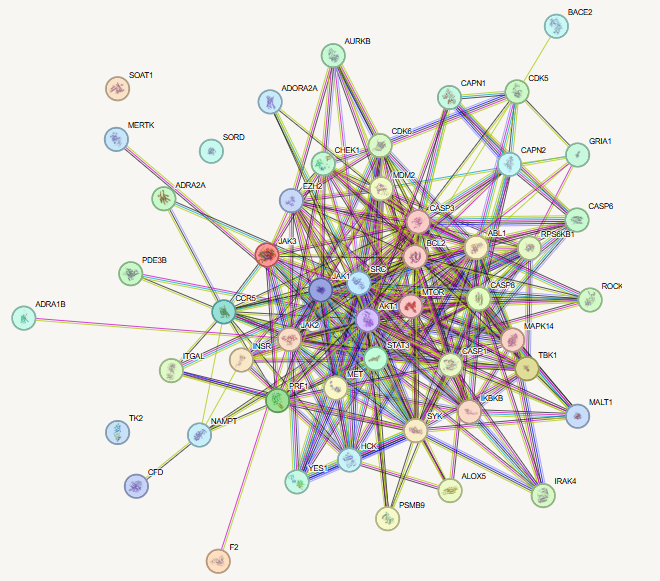

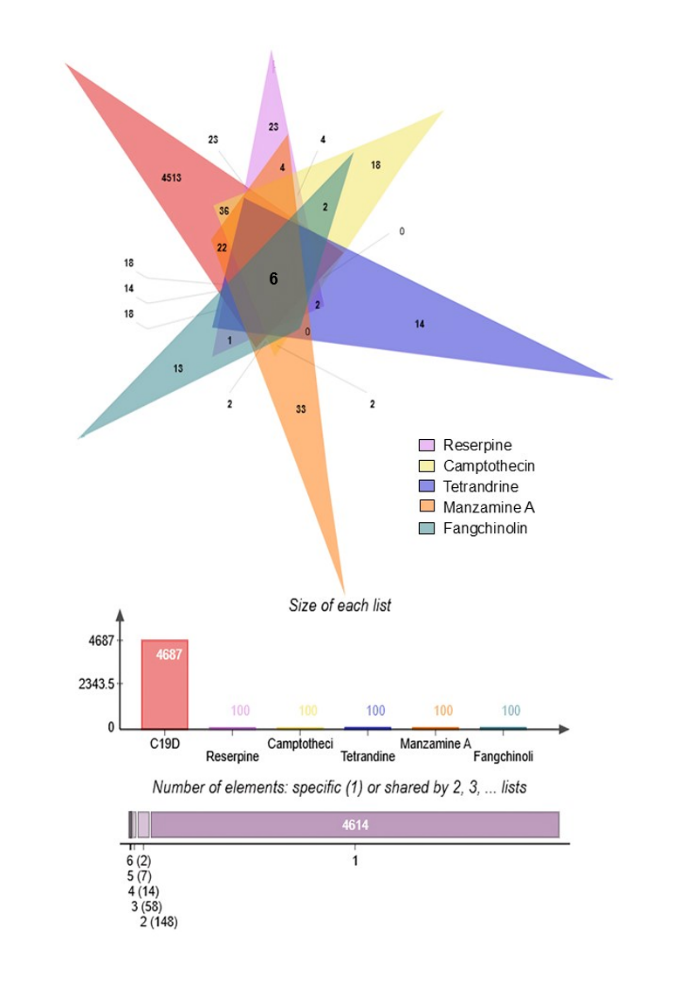
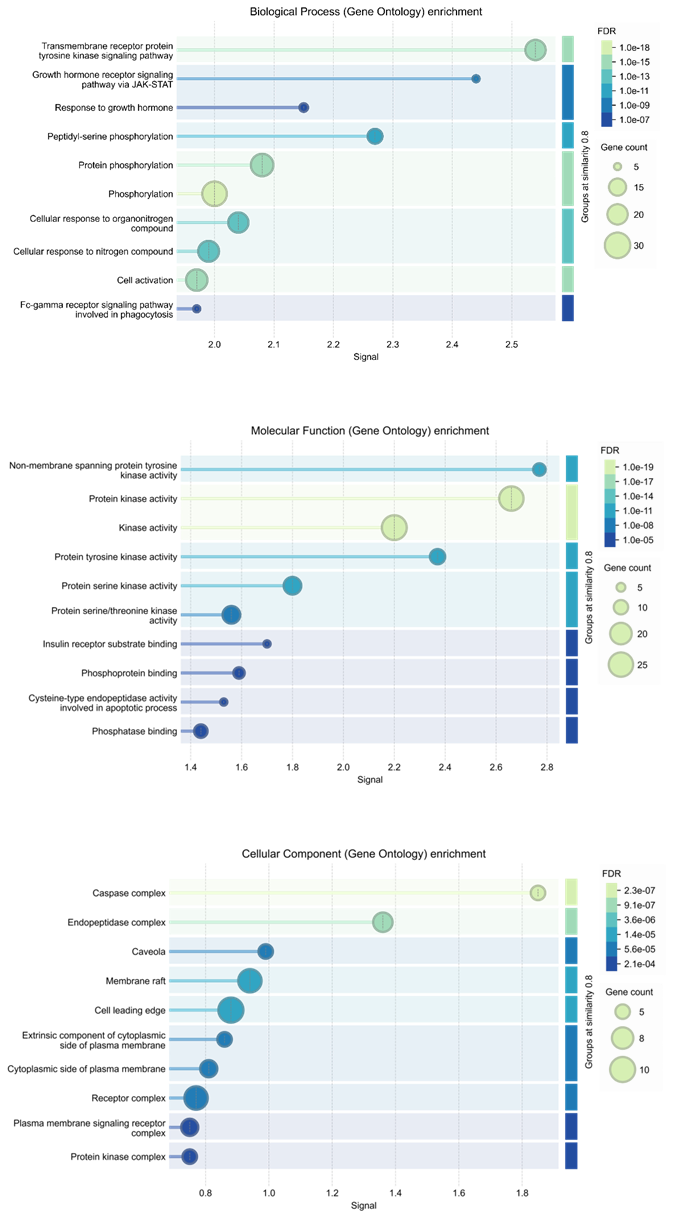
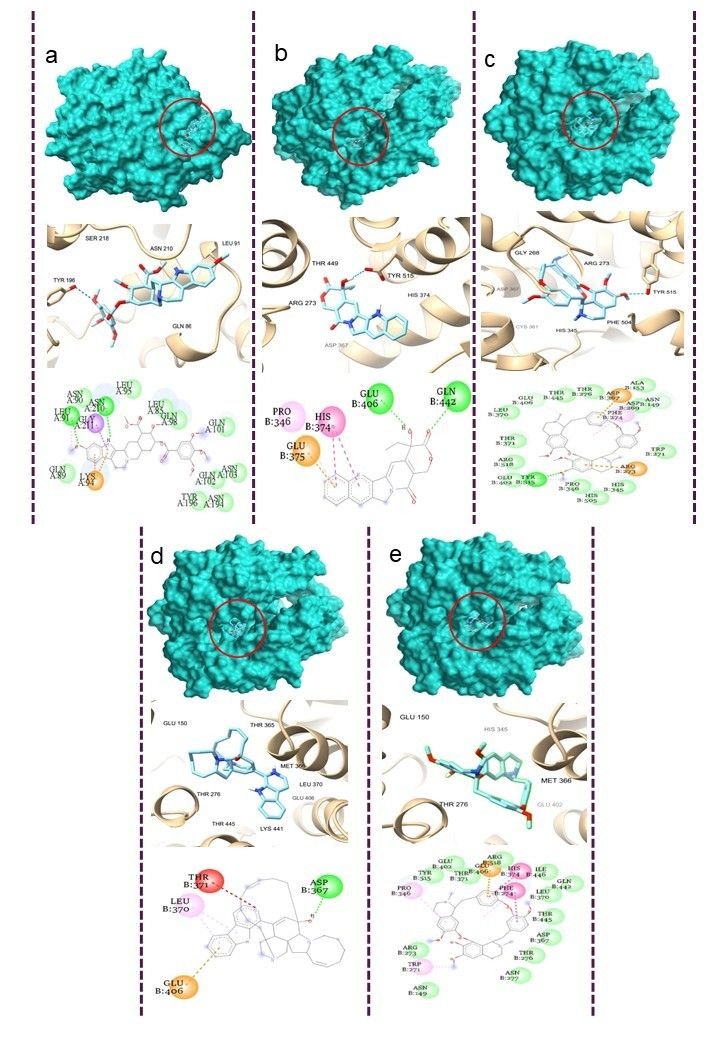
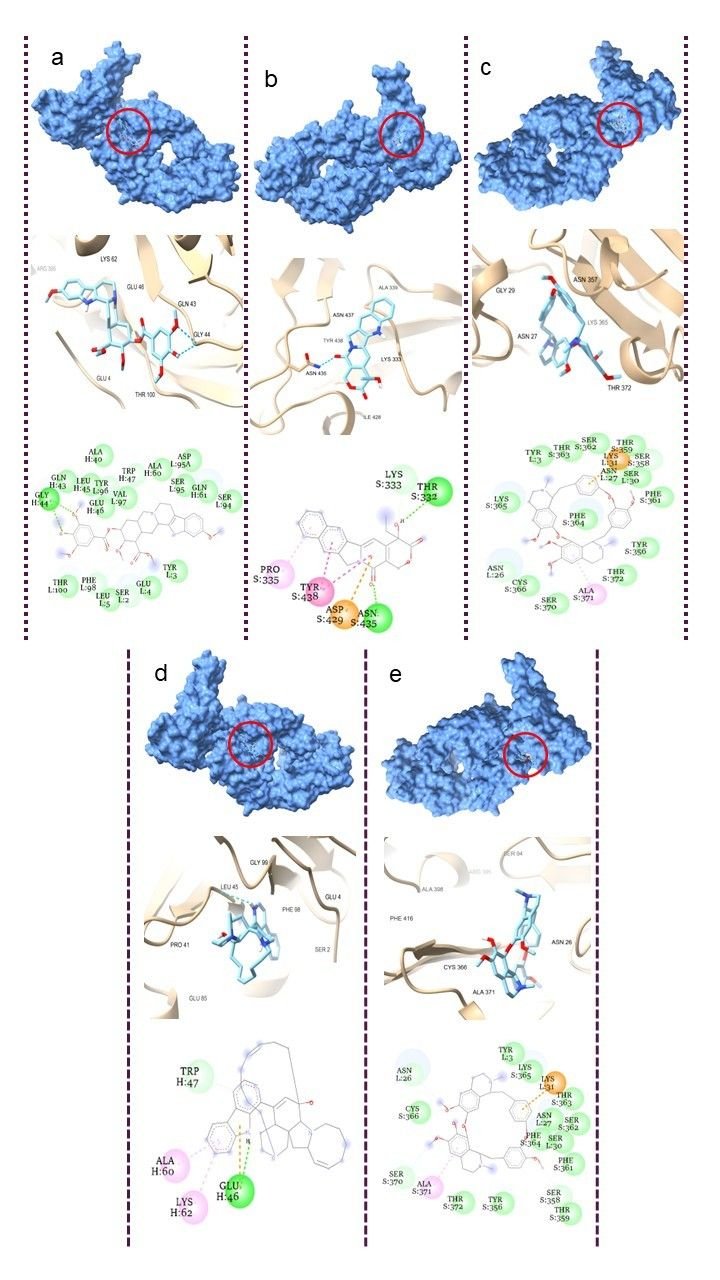
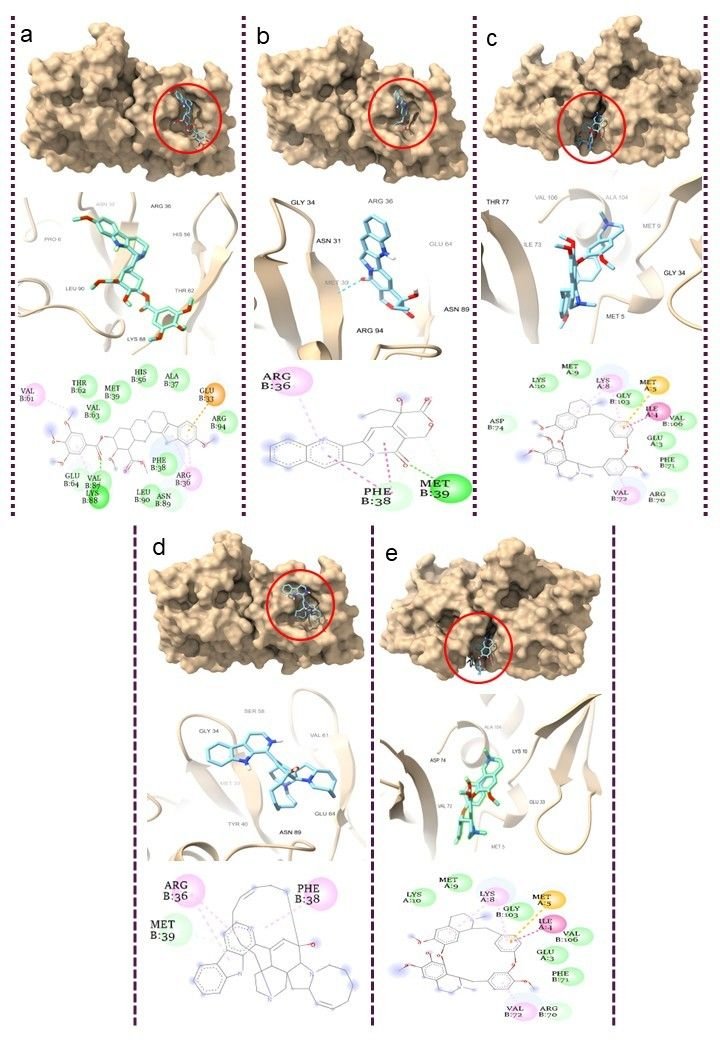
Molecular docking serves as an essential computational methodology for elucidating the interaction dynamics between prospective drug molecules and specific active or binding sites of target proteins, thereby facilitating rational drug design and the identification of high-affinity lead compounds (23). The strength of interaction between a ligand and the active sites of a target protein is primarily understood by analyzing hydrogen bonding networks and identifying key amino acid residues involved in these interactions (Figure 5). Six intersecting genes are found between disease-related targets and specific alkaloids-associated targets according to the venn diagram (Figure 6). A ligand demonstrating the highest binding affinity is often prioritized as a promising candidate for subsequent experimental validation and development (10). Biological process, molecular function and cellular enrichment of the intersecting genes between disease-associated genes and alkaloid targets through gene ontology study (Figure 7).
The CASTp server was employed to delineate and characterize the catalytic or active site regions within the target protein structure (24). Figure 8, presents the 3D structure of the proteins and showcases its key binding sites. For the protein structures 2AJF, 2DD8, and 2J98, the identified binding pocket volumes were measured as 228, 74, and 25 ų, respectively. The ideal ligand binding region was pinpointed at pocket 1 of each protein, with dimensions, 2AJF- volume 10786.289 ų, area 5562.820 ų; 2DD8- volume 3783.84 ų, area 1982.439 ų; 2J98- volume 38.568 ų, area 81.894 ų. To evaluate the structural quality of the protein, the PROCHECK server was used to generate a Ramachandran plot, which visualizes the distribution of backbone dihedral angles and distinguishes favourable from unfavourable conformational regions (Figure 9). A reliable and well-refined protein model is generally indicated when over 90% of its residues occupy energetically preferred regions (25).
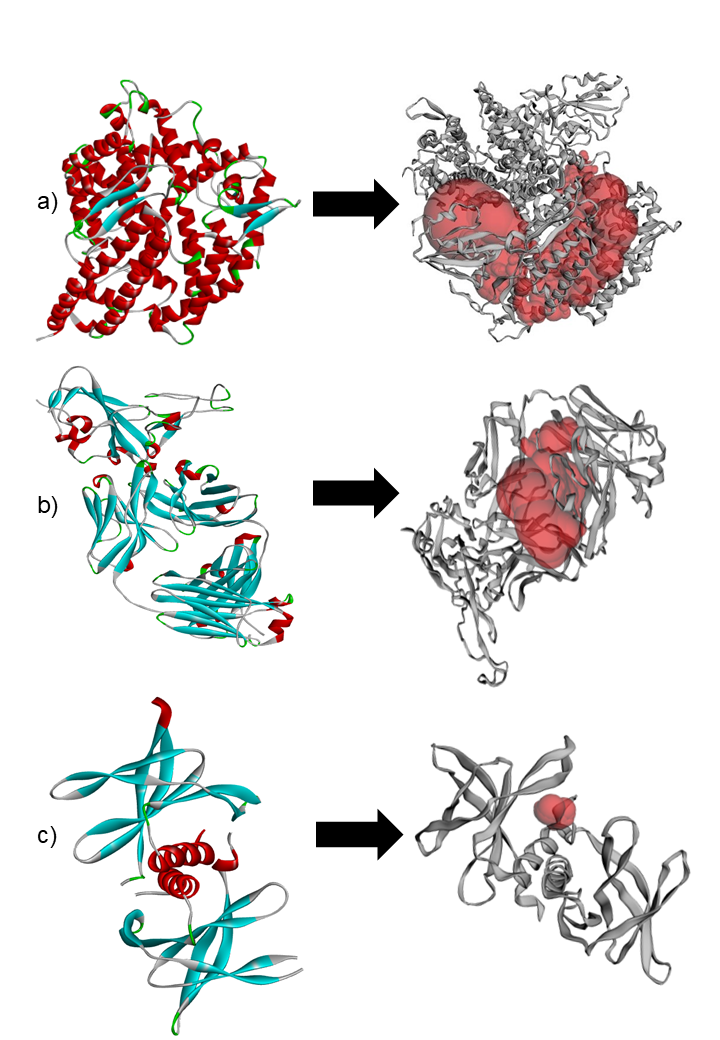
In this study, the target proteins demonstrated strong structural integrities, with 85.4% for 2AJF, 86.0% for 2DD8 and 92.0% for 2J98 proteins. Its residues located in the most favoured regions and only 0.4% occupying disallowed zones. This validation supports the accuracy of the proteins model and indicates reduced steric clashes through optimal φ and ψ torsion angle alignment, thereby enhancing its stereochemical stability for subsequent molecular docking analyses. With this goal, 5 plant-derived alkaloids, recognized for their antiviral activity, were selected for molecular docking against Human SARS-CoV virus proteins (PDB ID: 2AJF, 2DD8 and 2J98) (Figures 10-12).
All assessed phytocompounds shown strong binding affinities for the target protein, with binding energies ranging from -7.0 to -11.3 kcal/mol, according to the docking analysis (Supplemental Table 1). Lower binding energy values correspond to stronger and more stable ligand–protein interactions, reflecting higher inhibitory potential (24). With a binding affinity of -11.3 kcal/mol for 2AJF, -10.0 kcal/mol for 2DD8, and -9.1 kcal/mol for 2J98, Manzamine A proved to be the most effective inhibitor among the investigated compounds by establishing stable contacts with important residues. Hemanthamine's binding affinities with proteins 2AJF and 2DD8 were the lowest, at -7.0 kcal/mol and -6.8 kcal/mol, respectively.
Discussion
The molecular docking analysis in this study effectively used computational tools such as CASTp and PROCHECK to identify and validate key binding sites on SARS-CoV-2 proteins, supporting rational drug design. CASTp characterized protein surface topography by locating pockets, cavities, and functional binding regions and by measuring pocket volumes and surface areas, which influence ligand accessibility and binding affinity. These CASTp results are consistent with recent studies on protein surface mapping and underscore the tool’s importance for targeted drug-design efforts (16). PROCHECK Ramachandran plot analysis showing over 85% of residues in favoured regions indicates that the protein models are stereo chemically sound and suitable for docking (Figure 9). High model quality is essential because steric clashes and unfavourable torsion angles can compromise ligand-binding predictions. Ensuring structural integrity improves the reliability of simulated ligand–protein interactions, which is critical for antiviral drug discovery against SARS-CoV-2 variants (10).
Binding affinity results identify plant-derived alkaloids such as manzamine A as promising inhibitors, with docking energies as low as −11.3 kcal·mol⁻¹. These findings support the growing evidence that phytochemicals can act as antiviral agents by forming stable hydrogen-bonding networks with key viral residues, underscoring their potential relevance in drug-discovery pipelines (18). Network pharmacology analysis predicts interactions between alkaloid-associated genes and genes linked to SARS-CoV-2, supporting a multi‑target therapeutic strategy. Identified signalling molecules—including MTOR, STAT3, AKT1, SYK, CASP3, and JAK2—indicate the complex pathways these compounds may modulate. Integrating molecular docking with systems‑level analysis therefore helps elucidate the potential mechanisms of action for these alkaloids (Figure 5) (7).
Future studies should augment this computational framework with molecular dynamics simulations to evaluate ligand–binding stability and kinetics over time, and with biochemical assays to experimentally validate antiviral activity. Investigating synergistic effects of combined alkaloids and assessing their impact on host immune responses through multi‑omics integration could inform optimized therapeutics. Expanding compound libraries to include synthetic analogs and applying machine‑learning–guided screening would accelerate identification of leads with improved specificity and bioavailability.
Conclusion
Plant-derived alkaloids, known for their antiviral potential, were evaluated via in-silico molecular docking against SARS-CoV-2 proteins 2AJF, 2DD8, and 2J98 to assess binding affinities. Manzamine A exhibited the strongest inhibitory potential with binding energies of -11.3 kcal/mol for 2AJF, -10.0 kcal/mol for 2DD8, and -9.1 kcal/mol for 2J98, indicating stable interactions that could disrupt viral entry, replication, or assembly. These results align with prior studies on manzamine alkaloids' favourable binding to SARS-CoV-2 structures, highlighting their promise as natural drug scaffolds rooted in traditional medicine. However, docking predictions require experimental validation through in-vitro/in-vivo assays, pharmacokinetic profiling, and toxicity studies to confirm efficacy and safety. Structural optimizations and advanced formulations could further enhance Manzamine A's clinical viability against SARS-CoV-2 and emerging viruses.
Declarations
Acknowledgment
The authors sincerely thank all the staff and research scholars of the PG and Research Department of Botany, Kongunadu Arts and Science College, for their invaluable support.
Conflict of Interest
The authors declare no conflict of interest.
Data Availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Ethics Statement
Not applicable.
Funding Information
This work received no external funding.
Supplemental Material
<b>Supplementary Table 1</b> can be accessed in the following <a class="cursor-pointer" href="https://etflin.com/file/document/20260705040517_108433_2937b7cd.docx">link</a>.
References
- World Health Organization. (2025). Epidemiological assessment of SARS-CoV-2 variants XFG and NB.1.8.1. WHO.
- Baden LR, El Sahly HM, Essink B, Kotloff K, Frey S, Novak R, et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N Engl J Med. 2021;384(5):403-416. doi: https://doi.org/10.1056/nejmoa2035389
- Skowronski DM and De Serres G. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N Engl J Med. 2021;384(16):1576-1578. doi: https://doi.org/10.1056/nejmc2036242
- CDC (2021). Post-vaccination infections and transmission. U.S. Centers for Disease Control and Prevention.
- Yamin R, Ahmad I, Khalid H, Perveen A, Abbasi SW, Nishan U, et al. Identifying plant-derived antiviral alkaloids as dual inhibitors of SARS-CoV-2 main protease and spike glycoprotein through computational screening. Front. Pharmacol. 2024;15:1369659. doi: https://doi.org/10.3389/fphar.2024.1369659
- Prameela A, Radhakrishnan A, Krishnasamy T. Elucidating Neuropharmacological Implications of Vincetene: A Multi-target Computational Study on Ataxia, Encephalitis, and Meningitis. Prospect. Pharm. Sci. 2025;23(4):103-114. doi: https://doi.org/10.56782/pps.490
- Radhakrishnan A, Prameela A, Jayasree M, Habibulla AK. An exhaustive computational bioassessment of plant‐derived terpenoids as potential SARS‐CoV‐2 inhibitors. Vietnam Journal of Chemistry. 2026;1(1):5-8. doi: https://doi.org/10.1002/vjch.70136
- Kandwal S, Fayne D. Genetic conservation across SARS-CoV-2 non-structural proteins – Insights into possible targets for treatment of future viral outbreaks. Virology. 2023;581:97-115. doi: https://doi.org/10.1016/j.virol.2023.02.011
- Bhatia N, Thareja S. Simulation, 3D-QSAR, molecular docking and pharmacophore mapping studies on indole derivatives as aromatase inhibitors. Letters in Drug Design & Discovery. 2025;22(1):100007. doi: https://doi.org/10.1016/j.lddd.2025.100007
- Borquaye LS, Gasu EN, Ampomah GB, Kyei LK, Amarh MA, Mensah CN, et al. Alkaloids from Cryptolepis sanguinolenta as Potential Inhibitors of SARS‐CoV‐2 Viral Proteins: An In Silico Study. BioMed Research International. 2020;2020(1):5324560. doi: https://doi.org/10.1155/2020/5324560
- Shaikh, P. N. R. S. P. K. R. S. S. (2025). Rational Drug Design and Development: Trends and future directions. Zenodo (CERN European Organization for Nuclear Research). https://doi.org/10.5281/zenodo.17615793
- Prabakaran P, Gan J, Feng Y, Zhu Z, Choudhry V, Xiao X, et al. Structure of Severe Acute Respiratory Syndrome Coronavirus Receptor-binding Domain Complexed with Neutralizing Antibody. Journal of Biological Chemistry. 2006;281(23):15829-15836. doi: https://doi.org/10.1074/jbc.m600697200
- Li F, Li W, Farzan M, Harrison SC. Structure of SARS Coronavirus Spike Receptor-Binding Domain Complexed with Receptor. Science. 2005;309(5742):1864-1868. doi: https://doi.org/10.1126/science.1116480
- World Health Organization. (2023). COVID-19 vaccines: efficacy, safety, and adverse effects. WHO.
- Ponnusamy R, Moll R, Weimar T, Mesters JR, Hilgenfeld R. Variable Oligomerization Modes in Coronavirus Non-structural Protein 9. Journal of Molecular Biology. 2008;383(5):1081-1096. doi: https://doi.org/10.1016/j.jmb.2008.07.071
- Tian W, Chen C, Lei X, Zhao J, Liang J. CASTp 3.0: computed atlas of surface topography of proteins. Nucleic Acids Research. 2018;46(W1):W363-W367. doi: https://doi.org/10.1093/nar/gky473
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26(2):283-291. doi: https://doi.org/10.1107/s0021889892009944
- Abookleesh FL, Al-Anzi BS, Ullah A. Potential Antiviral Action of Alkaloids. Molecules. 2022;27(3):903. doi: https://doi.org/10.3390/molecules27030903
- Kim S, Chen J, Cheng T, Gindulyte A, He J, He S, et al. PubChem in 2021: new data content and improved web interfaces. Nucleic Acids Research. 2020;49(D1):D1388-D1395. doi: https://doi.org/10.1093/nar/gkaa971
- O'Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: An open chemical toolbox. J Cheminform. 2011;3(1):33-36. doi: https://doi.org/10.1186/1758-2946-3-33
- BIOVIA. (2025). Discovery Studio Visualizer (Version 2025). San Diego: Dassault Systèmes.
- Bardou P, Mariette J, Escudié F, Djemiel C, Klopp C. jvenn: an interactive Venn diagram viewer. BMC Bioinformatics. 2014;15(1):3-7. doi: https://doi.org/10.1186/1471-2105-15-293
- Pandey P, Rane JS, Chatterjee A, Kumar A, Khan R, Prakash A, et al. Targeting SARS-CoV-2 spike protein of COVID-19 with naturally occurring phytochemicals: an in silico study for drug development. Journal of Biomolecular Structure and Dynamics. 2020;39(16):6306-6316. doi: https://doi.org/10.1080/07391102.2020.1796811
- Prasanth DSNBK, Singh G, Panda SP, Achanti S, Soni H, Chaudhuri TK, et al. In Silico Screening of Plant-Derived Anti-virals from Shorea hemsleyana (King) King ex Foxw Against SARS CoV-2 Main Protease. Chemistry Africa. 2022;6(1):345-366. doi: https://doi.org/10.1007/s42250-022-00521-2
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26(2):283-291. doi: https://doi.org/10.1107/s0021889892009944