RESEARCH ARTICLE
Evaluation of an Ultra-Sensitive PCR Assay for Porcine Mitochondrial Cytochrome b Detection under Laboratory Conditions
Halal Science|Vol. 1, Issue 2, pp. 50-54 (2025)
CC BY 4.0-2025 Authors
Views
Downloads
Shares
Received
Sep 27, 2025Revised
Dec 25, 2025Accepted
Dec 27, 2025Published
Dec 31, 2025
Abstract
Food adulteration with porcine derivatives poses significant challenges to halal authentication and consumer protection. Sensitive molecular detection methods are therefore required to support food authenticity assessment. This study aimed to evaluate the analytical sensitivity of a PCR-based assay targeting porcine mitochondrial cytochrome b (cyt b) DNA. Genomic DNA was isolated from porcine intestine, adipose tissue, liver, blood, and muscle using the Wizard® Genomic DNA Purification Kit, quantified by spectrophotometry, and amplified using species-specific primers. Analytical sensitivity was assessed using stepwise serial dilutions of porcine DNA under controlled laboratory conditions. The extracted DNA showed acceptable purity for PCR amplification, with A260/A280 ratios within the expected range. PCR amplification produced detectable cyt b bands across a wide range of nominal DNA dilution levels, indicating high analytical sensitivity of the assay. The detection limit was determined operationally based on consistent visual band detection following agarose gel electrophoresis. These findings demonstrate the potential of the optimized PCR assay as a sensitive molecular tool for porcine DNA detection, with prospective applicability for halal authentication and food safety monitoring pending further validation in complex food matrices.
Introduction
Ensuring the safety and authenticity of food products is a critical public health and socio-religious concern (1). In Indonesia, where over 87% of the population identifies as Muslim, the assurance of halal food is both a legal requirement and a consumer right (2). However, rising domestic demand for animal protein, particularly beef, coupled with supply shortages, has created opportunities for food adulteration, including the fraudulent substitution of beef with pork (3). Such practices not only contravene Islamic dietary laws but also undermine consumer trust and market integrity (4). Reports of pork being disguised as beef or incorporated into processed meat products have raised widespread concern, highlighting the urgent need for sensitive, reliable, and standardized detection methods (5).
Traditional approaches to verifying meat authenticity such as morphological and biochemical tests are limited by their susceptibility to environmental factors, processing methods, and low discriminatory power (6). These constraints underscore the need for molecular biology-based techniques, which offer high specificity and sensitivity. Polymerase Chain Reaction (PCR) has been extensively applied for meat authentication and species identification, including both conventional end-point PCR and real-time PCR–based assays (7). Previous studies have demonstrated that PCR targeting mitochondrial DNA markers enables reliable detection of animal species even in highly processed food matrices; however, reported sensitivity levels and detection limits vary considerably depending on primer design, target gene, and analytical platform used (8). Among mitochondrial DNA markers, the cytochrome b (cyt b) gene has been widely used due to its evolutionary conservation across species and sufficient interspecies variability, making it an effective target for differentiating animal derived food components (9).
While real-time PCR offers higher analytical sensitivity and quantitative capability, its application is often constrained by higher operational costs, specialized instrumentation, and technical expertise. In many halal authentication laboratories, particularly in developing testing infrastructures, conventional PCR remains the most accessible and routinely implemented molecular method. Despite its widespread use, systematic evaluation of the analytical limit of detection (LOD) achievable by conventional PCR for pork DNA especially under controlled laboratory conditions emains limited in the context of halal food authentication. This lack of standardized sensitivity benchmarks may affect result interpretation and inter-laboratory comparability (10).
Therefore, this study was specifically designed to determine the analytical limit of detection (LOD) of pork mitochondrial DNA under controlled laboratory conditions using conventional PCR amplification of the cytochrome b (cyt b) gene. DNA was extracted from representative pig tissues, serially diluted to defined concentrations, and amplified using species-specific primers, followed by visualization through agarose gel electrophoresis (11). By establishing the minimum detectable DNA concentration using an end-point PCR approach, this study aims to provide a practical sensitivity benchmark for laboratories involved in routine halal authentication testing. Ultimately, this work supports the development of reliable, cost-effective molecular detection strategies to strengthen consumer protection and regulatory enforcement in predominantly Muslim markets.
Methodology
Study Design and Setting
This study was designed as a laboratory-based experimental investigation to evaluate the sensitivity and determine the limit of detection (LOD) of porcine mitochondrial DNA fragments encoding the cytochrome b (cyt b) gene using polymerase chain reaction (PCR). The research was conducted between February and March 2018 at the Genetics and Tissue Culture Laboratory, UIN Sunan Ampel, Surabaya, Indonesia, a facility equipped for molecular biology analysis relevant to halal authentication research.
Samples and Materials
Biological samples were obtained from fresh pig (Sus scrofa) tissues, including muscle (skeletal meat), liver, adipose tissue, intestine, and blood. Each tissue type was represented by a single biological sample used for DNA extraction and subsequent sensitivity testing. No biological replication was applied at the tissue level.
DNA extraction was performed using the Wizard® Genomic DNA Purification Kit (Promega, USA) following the manufacturer’s protocol with minor adjustments. The extracted DNA was eluted in 100 μL rehydration buffer and stored at 4 °C until further analysis.
DNA Quantification and Quality Assessment
DNA concentration and purity were measured spectrophotometrically (Biochrom Biodrop DUO, UK) at wavelengths of 260 nm and 280 nm. DNA purity was assessed using the A260/A280 ratio. As each tissue type was represented by a single extracted sample, the reported concentration and purity values reflect individual measurements and are presented descriptively without statistical analysis.
PCR Amplification
PCR amplification targeted a 149 bp fragment of the porcine mitochondrial cyt b gene. Reactions were carried out in a final volume of 25 μL, consisting of 2.5 μL template DNA, 12.5 μL GoTaq® Green Master Mix (Promega), 1 μL each of forward primer (Pork-F: 5’-ATGAAACATTGGAGTAGTCCTACTATTTACA-3’) and reverse primer (Pork-R: 5’-CTACGAGGTCTGTTCCGATATAAGG-3’) at 10 μM, and 8 μL nuclease-free water.
Amplification was performed using a Labnet MultiGene Optimax thermocycler under the following cycling conditions: initial denaturation at 98 °C for 2 min; 35 cycles of denaturation at 95 °C for 30 s, annealing at 61 °C for 30 s, and extension at 72 °C for 40 s; followed by a final extension at 72 °C for 3 min. Negative and specificity controls were included in each PCR run alongside the test samples to monitor contamination and cross-reactivity
Sensitivity and Limit of Detection Testing
To evaluate analytical sensitivity, porcine DNA extracts were subjected to stepwise ten-fold serial dilutions prepared from a quantified stock solution. Dilutions were generated sequentially (1: 10) using nuclease-free water, starting from the highest measurable DNA concentration down to the lowest nominal dilution level (10⁻⁶ pg). Each dilution step was prepared freshly from the immediately preceding dilution to minimize cumulative pipetting error.
The concentration values reported for each dilution level represent nominal dilution levels derived from the initial stock DNA concentration, rather than absolute DNA mass per PCR reaction. Given the extremely low DNA amounts at the highest dilution levels, no attempt was made to calculate the absolute number of target DNA molecules per reaction.
For PCR amplification, a fixed template volume of 2.5 µL from each dilution was added to the reaction mixture. Each dilution level was tested in triplicate to assess reproducibility of amplification at that nominal dilution level.
Operational Definition of Limit of Detection
In this study, the limit of detection (LOD) was operationally defined as the lowest nominal DNA dilution level that consistently produced a visible PCR amplification band of the expected size (149 bp) across all replicate reactions. Consistency was defined as the presence of a detectable band in three out of three PCR replicates at a given dilution level.
LOD determination was based on qualitative visual assessment of agarose gel electrophoresis results and should therefore be interpreted as an operational, gel-based detection limit rather than an absolute molecular detection threshold.
Gel Electrophoresis and Visualization
PCR products were separated on 2% agarose gels prepared in 0.5× TAE buffer and stained with Diamond™ Nucleic Acid Dye (Promega). Electrophoresis was performed at 50 V for 20 min. A 100 bp DNA ladder (BenchTop, Promega) was used as a molecular size reference. DNA bands were visualized using a UV transilluminator (Enduro GDS-1302) and digitally documented.
Data Analysis
LOD determination was based on qualitative and semi-quantitative evaluation of PCR results, defined by the presence or absence of the target band and relative band intensity across serial dilutions. No absolute DNA quantification was inferred from band intensity. DNA concentration and purity measurements were interpreted descriptively, consistent with the experimental design.
Result and Discussion
This study aimed to determine the analytical limit of detection (LOD) of porcine mitochondrial cytochrome b (cyt b) DNA using a PCR-based assay under controlled laboratory conditions (12).
DNA Isolation
Genomic DNA was extracted from porcine intestine, adipose tissue, liver, blood, and muscle obtained from Petemon Market, Surabaya. Approximately 50 mg of each tissue sample was processed using the Wizard® Genomic DNA Purification Kit (Promega, USA), which was selected for its proven efficiency, consistency, and reproducibility in isolating high-quality genomic DNA suitable for downstream PCR applications.
Briefly, tissue samples were mechanically homogenized in 600 µL of Nuclei Lysis Solution and incubated at 65 °C for 30 min to ensure complete cell lysis and inactivation of endogenous nucleases and other PCR-inhibitory enzymes. To remove RNA contaminants that could interfere with spectrophotometric measurements, 3 µL of RNase solution was added, followed by incubation at 37 °C for 30 min. Protein and salt residues were subsequently precipitated by the addition of 200 µL Protein Precipitation Solution, vortexed thoroughly, and incubated at –20 °C for 5 min. After centrifugation at 1300–1600 rpm for 4 min, the clarified supernatant was transferred to 600 µL isopropanol to precipitate genomic DNA. The resulting DNA pellet was washed with 70% ethanol (60 µL), air-dried to remove residual ethanol, and rehydrated in 100 µL DNA Rehydration Solution (or TE buffer).
DNA concentration and purity were assessed using a Biochrom Biodrop-DUO spectrophotometer by measuring absorbance at 260 nm and 280 nm. The A260/A280 ratios obtained ranged from 1.77 to 1.96, indicating that the extracted DNA exhibited acceptable purity for PCR amplification. Among the tissues analyzed, liver samples yielded the highest DNA concentration (19.89 µg/mL), whereas blood samples showed the lowest concentration (0.61 µg/mL), which may be attributed to differences in nucleated cell content and tissue-specific extraction efficiency (Table 1).
| No | Sample | A260 | A280 | Concentration (µg/mL) | A260/A280 Ratio |
|---|---|---|---|---|---|
| 1 | Intestine | 0.238 | 0.143 | 10.86 | 1.777 |
| 2 | Adipose | 0.054 | 0.046 | 0.887 | 1.821 |
| 3 | Liver | 0.430 | 0.249 | 19.89 | 1.835 |
| 4 | Blood | 0.008 | 0.002 | 0.611 | 1.964 |
| 5 | Muscle | 0.047 | 0.028 | 2.097 | 1.808 |
Although the intestine sample showed a slightly lower A260/A280 ratio (1.77), this value remained within the acceptable range for downstream PCR analysis, indicating minimal protein contamination. Variations in DNA yield among tissue types likely reflect inherent biological differences under standardized extraction conditions.
Because each tissue type was represented by a single extracted biological sample, DNA concentration and purity values are presented as single representative measurements and were interpreted descriptively without statistical analysis or inference of biological variability. These values are not intended to reflect population-level variation or support quantitative comparisons between tissue types. The measurements were intended solely to confirm the suitability and analytical adequacy of the extracted DNA for subsequent PCR amplification and sensitivity testing.
PCR Amplification and Sensitivity Analysis
PCR amplification targeting a 149 bp fragment of the porcine mitochondrial cytochrome b (cyt b) gene was successfully achieved using species-specific primers. Mitochondrial DNA was selected as the target due to its high copy number and relative resistance to degradation, which enhances analytical sensitivity.
Serial dilutions of porcine DNA were prepared to assess assay sensitivity. DNA input was expressed as the nominal amount of DNA per PCR reaction, calculated based on the dilution concentration and a template volume of 2.5 µL. Each dilution level was tested in triplicate PCR reactions to evaluate amplification consistency.
Strong and distinct amplification bands were consistently observed at higher DNA input levels. As the DNA input decreased, band intensity gradually diminished, reflecting reduced template availability.
Gel Electrophoresis and Visualization
Gel electrophoresis was performed to separate and visualize PCR products based on DNA fragment size and charge (13). DNA is negatively charged and migrates toward the anode in an electric field, with migration speed determined by fragment size, gel concentration, and applied voltage (14).
A 2% agarose gel was prepared in 0.5× TAE buffer, supplemented with 10 µL Diamond™ Nucleic Acid Dye (Promega) for DNA visualization. Samples were mixed with 1 µL loading dye (containing bromophenol blue and glycerol) to allow tracking of migration and to ensure proper sample deposition in wells. A 100 bp DNA ladder was loaded in the first lane, followed by 5 µL of DNA samples and a negative control (Psidium guajava L. ) in the last lane.
Electrophoresis was carried out at 50 V for 100 min. DNA fragments were visualized under UV illumination, where band position reflected fragment size and relative migration. The gel electrophoresis procedure is shown in Figure 1.

Visualization of PCR Products and Determination of LOD
The quality of PCR amplification was assessed by agarose gel electrophoresis followed by UV transillumination. PCR products were evaluated across a series of stepwise ten-fold serial dilutions of porcine DNA, ranging from high nominal DNA input levels to the lowest dilution tested.
Clear and distinct amplification bands of the expected size (149 bp) were consistently observed at higher nominal dilution levels, indicating efficient amplification of the porcine mitochondrial cyt b gene. As the DNA input decreased, band intensity gradually diminished, reflecting reduced template availability. At the lowest dilution levels, faint bands were occasionally observed, suggesting that amplification was approaching the analytical detection limit of the assay.
It should be noted that the detection of faint bands at extreme dilution levels represents qualitative visual detectability under optimized laboratory conditions using agarose gel electrophoresis. From a molecular perspective, such nominal DNA inputs may statistically correspond to an average target copy number at or below one molecule per PCR reaction (15, 16), and therefore the resulting amplification signals should be interpreted with caution and should not be considered evidence of consistent single-molecule detection.
Accordingly, the limit of detection (LOD) in this study is defined as an operational, gel-based detection limit determined by consistent visual detection of the target band across replicate reactions, rather than an absolute molecular threshold. The reported LOD thus reflects practical analytical sensitivity under controlled experimental conditions.
At the defined LOD level, amplification was consistently observed in 3 out of 3 PCR replicates, whereas at lower nominal dilution levels amplification became inconsistent or was not observed. This consistency defines the LOD as a practical, method-dependent gel-based detection limit under experimental conditions.
Figure 2 illustrates representative agarose gel electrophoresis results obtained from serially diluted porcine DNA templates. Amplification signals were observed across a wide range of nominal dilution levels, supporting the high analytical sensitivity of the PCR assay (17, 18). However, direct comparison of LOD values with previous studies should be made cautiously, as differences in experimental design, DNA preparation, PCR chemistry, cycling conditions, and visualization methods can substantially influence apparent detection limits.
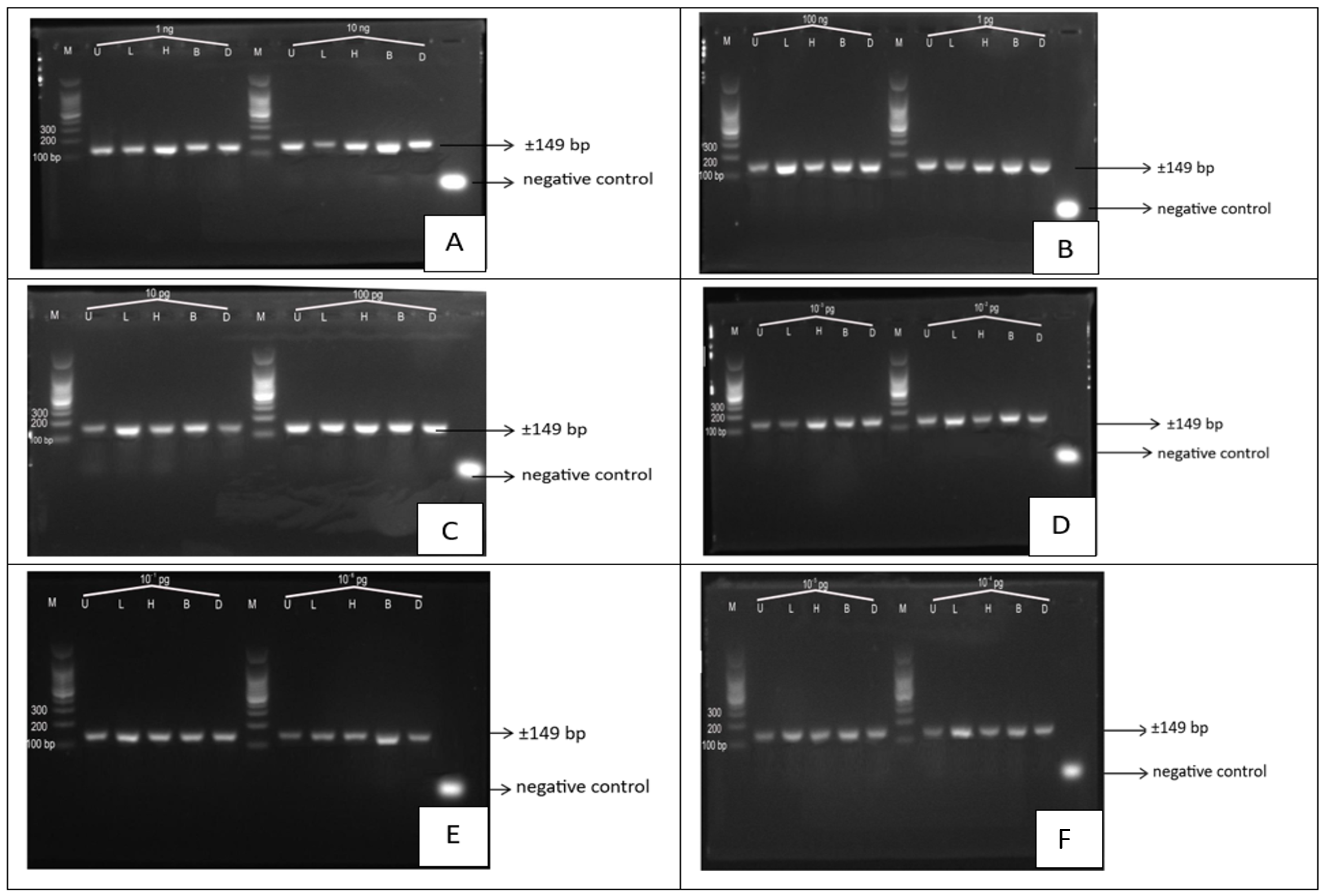
The analytical sensitivity observed in this study was achieved using purified porcine DNA under laboratory-controlled conditions. As the experiments did not involve complex food matrices or mixed-species samples, the application of this assay for halal authentication should be regarded as prospective (19, 20). Further validation using processed food products and real-world samples is required to establish robustness and applicability for routine halal certification.
Comparison with Previous Studies
The lower nominal detection limit observed in this study compared with previous reports may be influenced by differences in experimental conditions, including DNA purity, primer design, PCR chemistry, cycling parameters, and visualization methods. Direct comparison of LOD values across studies should therefore be interpreted with caution, as methodological and platform-specific factors can substantially affect apparent sensitivity (17).
Halal Authentication Claim
The high analytical sensitivity observed in this study represents performance under controlled laboratory conditions using purified porcine DNA. Because the experiments did not involve complex food matrices or mixed-species samples, the applicability of this assay for halal authentication should be regarded as prospective. Further validation using processed food products and real-world samples is required before routine application in halal certification workflows.
Implications and Study Limitations
The high sensitivity demonstrated in this study reflects analytical sensitivity obtained using purified porcine DNA under controlled laboratory conditions. As the experiments were conducted exclusively on DNA extracted from pure porcine tissues, the performance of this assay in complex food matrices, processed products, or mixed-species samples was not evaluated.
Accordingly, the application of this PCR assay for halal authentication should be regarded as a prospective use. Further studies incorporating real food products and mixed matrices are required to validate the robustness, reproducibility, and practical applicability of this method for routine halal certification and food authenticity testing.
Conclusion
This study demonstrates the development of a PCR-based assay with high analytical sensitivity for the detection of porcine mitochondrial cytochrome b (cyt b) DNA under controlled laboratory conditions. The combination of efficient DNA extraction, the use of species-specific primers, and optimized PCR parameters enabled reliable amplification across a broad range of nominal DNA dilution levels, as assessed by gel-based visualization. In this study, the limit of detection was defined operationally based on consistent visual detection of the target amplicon rather than on absolute molecular quantification or single-copy detection.
While the results indicate strong analytical performance, further studies incorporating processed food products, complex food matrices, and mixed-species samples are required to evaluate the robustness, reproducibility, and practical applicability of this assay before it can be applied for routine halal authentication and food safety monitoring.
Declarations
Conflict of Interest
The authors declare no conflicting interest.
Data Availability
All data generated or analyzed during this study are included in this published article.
Ethics Statement
Ethical approval was not required for this study.
Funding Information
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
References
- Fauzi AHM, Yahya H, Yahaya N, Hassan MS, Yahya HN. The juxtaposition of food safety and halal regulations. Int J Relig. 2024 Aug 30;5(11):8138–8146.
- Durrotul H, Riset FP, Pengembangan D, Halal P. Sertifikasi halal di Indonesia: sejarah, perkembangan, dan implementasi. J Halal Prod Res. 2019;2(2):68–78.
- Siswara HN, Erwanto Y, Suryanto E. Study of meat species adulteration in Indonesian commercial beef meatballs related to halal law implementation. Front Sustain Food Syst. 2022 Jul 5;6:882031.
- Tahir P, Muslih M. Halal and safe food in Islamic law. Batulis Civil Law Rev. 2023 May 2;4(1):37.
- Sugiana FA, Widyowa H, Warisman MA, Suryani, Desriani. Low cost and comprehensive pork detection in processed food products with different food matrices. Indones J Biotechnol. 2018;23(1):21–27.
- Fathima AM, Rahmawati L, Windarsih A, Suratno. Advanced halal authentication methods and technology for addressing non-compliance concerns in halal meat and meat products supply chain: a review. Food Sci Anim Resour. 2024;44(6):1195–1212.
- Aina GQ, Rohman A, Erwanto Y. Wild boar-specific PCR assay and sequence analysis based on mitochondrial cytochrome B gene for halal authentication studies. Indones J Chem. 2020;20(2):483–492.
- Malau J, Kasasiah A, Zahra AA, Irwansyah SL, et al. Development of species-specific primers targeting mitochondrial Cyt b gene for porcine DNA detection in halal authentication via polymerase chain reaction (PCR). J Pembelajaran Biol Nukleus [Internet]. 2025;11(2):352–374.
- Vaithiyanathan S, Vishnuraj MR, Reddy GN, Srinivas C. Authentication of camel meat using species-specific PCR and PCR-RFLP. J Food Sci Technol. 2021 Oct;58(10):3882–3889.
- Rahma AA, Meilani ND, Sulistiawati, Ainaputri AS, Damara DS, Malau J. Development of a gelatin-based genomic reference material for halal authentication using real-time PCR. Sci Technol Indones. 2025 Jan 1;10(1):27–42.
- Zabidi AR, Fauzi FN, Abd Razak FN, Rosli D, Jamil MZM, Wan Ibrahim WK, et al. Screening porcine DNA in collagen cream cosmetic products. Food Res. 2020;4(1):151–156.
- Khayyira AS, Estepane VM, Malik A. Rapid PCR-based detection optimization of porcine DNA in gelatin capsule shell. Int J Appl Pharm. 2018;10(6):217–223.
- Ramlan RR, Harnelly, Fitri L. DNA extraction and PCR optimization of Coffea arabica L. and Coffea canephora Pierre ex A. Froehner. J Penelit Pendidik IPA. 2024 Aug 25;10(Special Issue):53–58.
- Hutahaean S. Penuntun praktikum bioteknologi. In: Prosiding Seminar Nasional Biologi [Internet]. 2014.
- Meki IK, Ahn KB, Dundon WG, Settypalli TBK, Leth C, Steinrigl A, et al. Novel multiplex family-wide PCR and Nanopore sequencing of amplicons (FP-NSA) approach for surveillance of influenza- and coronaviruses in humans and animals. Genome Med. 2025 Oct 17;17(1):123.
- Peng X, Yan M, Yang H, Zhen L, Wei L, Xu H. Fragment-specific quantification of 5hmC by qPCR via a combination of enzymatic digestion and deamination: extreme specificity, high sensitivity, and clinical applicability. Anal Chem. 2025 Feb 4;97(4):2186–2194.
- Parwati I, Chaidir L, Yunus M, Montain MM, Budhiarko D, Selasih SF, et al. Evaluation of a real-time PCR assay performance to detect Mycobacterium tuberculosis, rifampicin, and isoniazid resistance in sputum specimens: a multicenter study in two major cities of Indonesia. Front Microbiol. 2024 May 10;15:1–8.
- Park MY, Lee KH, Yang YJ, Seo JY, Lee JK, Jang JH, et al. Development of a multiplex real-time PCR assay for the rapid detection of the Asian-type DEL. J Mol Diagn. 2025 Sep;27(9):841–849.
- Rahma AA, Meilani ND, Sulistiawati, Ainaputri AS, Damara DS, Malau J. Development of a gelatin-based genomic reference material for halal authentication using real-time PCR. Sci Technol Indones. 2025 Jan 1;10(1):27–42.
- Abdullah Sani MS, Basuki CDP, Zainal R. Evaluating PCR and ELISA for porcine detection in collagen-based products for halal authentication. Halalsphere. 2025 Jul 31;5(2):8–18.