
RESEARCH ARTICLE
The Effect of Poloxamer 188 on the Solubility and Dissolution Behaviors of Piroxicam-PEG 4000 Solid Dispersions
Academic Editor: Adeleye Ademola Olutayo
Sciences of Pharmacy|Vol. 4, Issue 4, pp. 261-269 (2025)
CC BY 4.0-2025 Authors
Views
Downloads
Shares
Received
Jul 27, 2025Revised
Sep 27, 2025Accepted
Oct 10, 2025Published
Nov 6, 2025
Abstract
Piroxicam (PRX), a non-steroidal anti-inflammatory drug (NSAID), is classified as a biopharmaceutical classification system class II (high permeability and low solubility), which limits its bioavailability. Enhancement of the dissolution rate is a key strategy to enhance the absorption. Solid dispersion systems, particularly when combined with amphiphilic multiple co-block polymers, offer a promising approach to address this challenge. This study aimed to investigate the effect of Poloxamer 188 (P188) and the solid dispersion technique on the solubility and dissolution rate of PRX. Polyethylene glycol (PEG) 4000-based solid dispersions containing PRX were prepared using varying concentrations of Poloxamer 188 surfactant through the fusion method. The solid dispersions were evaluated for saturated solubility in water for 24 hours. Selected formulations were further characterized using thermal analysis and vibrational spectroscopy. The optimized solid dispersion formulation was filled into capsules, and a dissolution assay was carried out to compare its performance with that of pure PRX capsules. The optimized formula, comprising 3% P188 and PEG4000, demonstrated a significant enhancement in saturation solubility parameters (p < 0.05), specifically supersaturation degree, precipitation rate, and supersaturation profile. Additionally, dissolution testing showed a 22.22% increase in the dissolution rate of the PRX solid dispersion capsules compared to pure PRX capsules. In conclusion, P188-based solid dispersion containing PRX enhanced the solubility and dissolution rate, potentially improving therapeutic efficacy.
Introduction
Low drug bioavailability is a common challenge in drug delivery systems, often resulting from factors such as poor water solubility and slow dissolution rates (1). Approximately 60-70% of potential drug candidates are classified as biopharmaceutical classification system (BCS) class II, which includes drugs with high permeability but low solubility (2). Piroxicam (PRX), a widely prescribed non-steroidal anti-inflammatory drug (NSAID) used to treat conditions such as rheumatoid arthritis and osteoarthritis, is an example of a BCS Class II drug (3, 4). It is practically insoluble in water, with a solubility value of 0.023 mg/mL (5). Low solubility can significantly affect a drug’s therapeutic efficacy due to limited gastrointestinal absorption and reduced bioavailability (6).
Various formulation strategies have been investigated to overcome the solubility limitations of PRX. These include solid dispersions with different polymeric carriers (7, 8), nanosuspensions to increase surface area and dissolution rate (9, 10), and inclusion complexes with cyclodextrins to improve aqueous solubility (11, 12). While these approaches have shown promise, they also present challenges, such as complex manufacturing processes, stability concerns, and limited scalability for industrial applications.
Solid dispersion remains a practical and widely applicable strategy to enhance drug solubility and bioavailability by dispersing the drug in a hydrophilic matrix molecularly (13). This approach has been demonstrated to enhance solubility, increase dissolution rates, and prevent drug recrystallization or agglomeration (14-16). The development of solid dispersions requires consideration of several factors, including the type and concentration of the carrier. The drug-to-carrier ratio influences drug release, while the selection of carrier type depends on its physicochemical properties, such as hygroscopicity and water solubility (17). One widely used carrier is PEG 4000, a water-soluble, non-hygroscopic, and non-toxic polymer (18). Previous studies have demonstrated that using PEG 4000 in solid dispersions of PRX enhances its dissolution rate; however, the improvement has been modest and inconsistent (7, 19).
Additionally, to further improve PRX solubility, surfactants are increasingly incorporated into solid dispersion systems to enhance solubility and stability. Surfactants enhance the wettability of the drug, prevent precipitation, and promote dissolution (20). For instance, Poloxamer 188 (P188), an amphiphilic co-block polymer, has been shown to improve the solubility of boswellic acid compounds (21), and may similarly benefit piroxicam formulations. However, despite the individual use of PEG- or Poloxamer-based systems, no study to date has systematically combined PEG 4000 with P188 in PRX solid dispersions, optimized their ratios using the melting method, and subsequently evaluated both physicochemical properties and dissolution performance in capsule dosage form.
This study aimed to fill this gap by developing and optimizing the solubility and dissolution rate of PRX by preparing a solid dispersion using the melting method, chosen for its simplicity, solvent-free nature, and suitability for materials with low melting points (22). The solid dispersion system was developed by combining PEG 4000 with various concentrations of P188, and its performance was assessed through solubility testing. The formulation exhibiting the best solubility was selected for further characterization using differential scanning calorimetry (DSC) and fourier transform infrared spectroscopy (FTIR). The optimized formulation was then encapsulated, and its dissolution profile was compared to that of pure PRX capsules.
Experimental Section
Materials
The materials used in this research include PRX (PT Kimia Farma, Batch No. 0000086681), PEG 4000 (The Dow Chemical Company, CAS No. 25322-68-3), P188 (Merck, CAS No. 137065-10-00), sodium starch glycolate (Sigachi Industries PVT. LTD, Batch No. SM014/18), magnesium stearate, lactose (Grande Custom Ingredients Group, Lot No. 190920020410), sodium chloride (Merck, Lot No. K49469804), methanol (PT Smart Lab, Batch No. 160119001), hydrochloric acid (PT Smart Lab, Batch No. 080221001), and size two gelatine-based capsule shells.
Preparation of PRX-PEG 4000-Poloxamer 188 Solid Dispersion
Solid dispersions were prepared using the melting method, with a PRX: PEG 4000 weight ratio of 1: 2 (8). Briefly, 1.1 g of PEG 4000 was melted in a water bath at 80 °C until a clear, homogeneous solution was obtained. Subsequently, 2.2 g of PRX was added to the PEG 4000 solution, and the mixture was stirred continuously until a uniform, transparent solution was achieved, ensuring complete dissolution and even distribution of the drug within the polymeric matrix.
To prepare Formula 1, Formula 2, Formula 3, and Formula 4, poloxamer 188 was incorporated at concentrations of 0%, 1%, 3%, and 5%, respectively. These concentration levels were selected based on preliminary screening results, which demonstrated that concentrations within this range effectively enhanced drug wettability and dispersion while avoiding undesirable processing issues such as foaming, stickiness, or phase separation. The resultant mixture was then cooled in an ice bath under constant stirring until a solid mass was formed. The solid mass was placed in a desiccator for 24 h to ensure complete drying and removal of residual moisture. Following this, the dried solid was finely ground and sieved through a 60-mesh sieve to obtain a uniform particle size distribution. The resulting powder was stored in a desiccator until further analysis (23).
Characterization of PRX-PEG 4000-Poloxamer 188 Solid Dispersion
Percentage Yield
The percentage yield of the solid dispersion was determined by comparing the weight of the resulting solid dispersion powder (before sieving) with the total initial weight of the constituent materials. The yield was calculated using the formula in Eq. 1 (7).
Solubility Study
The solubility of the PRX solid dispersion was evaluated by adding an excess amount of solid dispersion powder to 8 mL of distilled water. The mixture was stirred continuously using a magnetic stirrer at a constant speed for 24 h. At predetermined time intervals (1, 3, 5, 7, 10, 15, 30, 45, 60, 120, 240, and 1440 min), 500 µL of the solution was withdrawn and centrifuged (24, 25). The supernatant was collected (100 µL) and diluted with 3 mL of distilled water. The sample was then analyzed using a UV spectrophotometer at 354 nm. The solubility profile was constructed to identify the optimal solid dispersion formula.
Fourier Transform Infrared Spectroscopy (FTIR)
FTIR analysis using KBr was performed to investigate potential interactions between the drug and carrier in the PRX-PEG 4000-Poloxamer 188 solid dispersion, as well as to detect the presence of any crystalline structures (26). The FTIR spectra of pure PRX, PEG 4000, poloxamer 188, and the PRX-PEG 4000-Poloxamer 188 solid dispersion were compared. Each sample was finely ground and mixed with potassium bromide to form a disc, which was compressed under a pressure of 6000 kg/cm² (27). The spectra data were recorded in the range of 4000–400 cm⁻¹ (7).
Differential Scanning Calorimetry (DSC)
DSC was employed as a thermal analysis technique to assess the stability of the solid dispersion system, focusing on polymorphic transitions, molecular mobility, crystallization, and drug-polymer miscibility (28). A 20 mg sample was weighed and placed in a sealed aluminum pan, then heated from 25 °C to 400 °C at a rate of 10 °C/min (29).
Preparation of PRX Solid Dispersion Capsules
Capsules containing PRX-PEG4000-P188 solid dispersions were prepared in 100-piece batches using size two capsules. The preparation process began by accurately weighing the required amounts of each ingredient to ensure formulation consistency and reproducibility. The solid dispersion of PRX-PEG4000-P188 equivalent to 10 mg, lactose (adjusted to obtain a total capsule formulation of 250 mg), and sodium starch glycolate (4%) was thoroughly mixed until homogeneous. Magnesium stearate (0.5%) was then added, and those components were mixed homogeneously for several min to ensure uniform lubrication and powder flow. The final mixture was encapsulated using a capsule filling machine under controlled environmental conditions. Afterwards, the filled capsules were carefully cleaned to remove any dust residue from the manufacturing process and stored in airtight containers for further evaluation.
Evaluation of Physicochemical Properties of PRX Solid Dispersion Capsules
Angle of Repose
The angle of repose test is an indirect method used to assess the flow properties of a powder preparation. The study is performed by allowing the powder to flow through a funnel from a specified height onto a horizontal surface (30). The angle of repose is then calculated from the height and diameter of the resulting cone, measured using a vernier calliper. The angle is determined using the formula in Eq. 2.
Uniformity of Weight
The weight uniformity test is used to evaluate the variation in capsule weight, which is directly related to dosage consistency. The test involves weighing one capsule, removing its contents, and then reweighing the capsule shell to determine the weight of the contents. This process is repeated for the remaining 19 capsules. Capsules are considered to meet the requirements if the average content weight exceeds 120 mg, with no more than two capsules deviating by more than 7.5% from the average weight and no single capsule deviating by more than 15% from the average weight (31).
Disintegration Time
The disintegration time test is conducted by placing one capsule into each of the six tubes of the disintegration tester basket. The medium used is water maintained at 37 ± 2 °C. The basket is then oscillated up and down in the medium at a frequency of 29–32 cycles per minute. Capsules are expected to disintegrate completely, except for the capsule shell. If one or two capsules fail to disintegrate fully, the test is repeated with an additional 12 capsules. At least 16 of the 18 capsules must disintegrate completely for the test to pass (32).
Validation of PRX Analytical Method
The validation of the analytical method was performed to assess accuracy, precision, and the potential for placebo interference. Accuracy and precision were evaluated by preparing three different concentrations of PRX solution in the dissolution medium (artificial gastric fluid without pepsin, pH 1.2), specifically 3.0, 5.0, and 15.0 µg/mL. Each concentration was analyzed in triplicate. Accuracy was determined by calculating the recovery, and precision was assessed by the relative standard deviation (RSD). Placebo interference was evaluated by comparing the absorbance of a solution containing the excipients used in the capsule with the PRX solution in the dissolution medium. Both solutions were analyzed at a wavelength of 354 nm, and the percentage of placebo interference was then calculated (32).
Dissolution Study
Artificial gastric fluid without pepsin (pH 1.2) was applied as dissolution medium. To prepare the dissolution medium, 2.0 g of NaCl was dissolved in 7.0 mL of HCl 37%, and distilled water was added to achieve a final volume of 1000 mL. A total of 900 mL of the dissolution medium was placed in the dissolution chamber, which was then set at 37 °C. The solid dispersion of PRX-PEG 4000-P188 was placed in the Type I dissolution tester, and the dissolution test was performed at a rotation and temperature of 50 rpm and 37 °C, respectively. Dissolution samples of 10 mL were withdrawn at 5, 10, 20, 30, 45, and 60 min. Furthermore, an equal volume of fresh medium at 37 °C was added to the chamber to maintain the volume. The absorbance of the samples was scanned using a UV-Vis spectrophotometer (Thermo Genesys S150; Waltham, MA) at a wavelength of 354 nm. The absorbance values were then used in the linear regression equation to calculate the concentration of PRX. The PRX-PEG 4000-P188 solid dispersion must dissolve at least 75% within 45 min to meet the criteria (32). The dissolution profiles of the solid dispersion capsules were compared to those of pure PRX capsules.
Statistical Analysis
All experiments were performed in triplicate, and the results are expressed as mean ± standard deviation (SD). Data were first evaluated for normality using the Shapiro–Wilk test and for homogeneity of variances using Levene’s test. When the data were normally distributed and homogeneous, statistical analysis was performed using one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test for pairwise comparisons. For normally distributed but non-homogeneous data, the Games–Howell post hoc test was applied. In cases where the data were not normally distributed, the Kruskal–Wallis test was employed. A p-value of < 0.05 was considered statistically significant. Data analysis was conducted using IBM SPSS Statistics version 22. For the dissolution study, the similarity factor (F2) was calculated to compare the dissolution profile of the optimized solid dispersion capsule with that of pure PRX capsules. An F2 value between 50 and 100 was considered to indicate similarity between the two dissolution profiles.
Results and Discussion
Characterization of PRX-PEG 4000-Poloxamer 188 Solid Dispersion
Percentage Yield
The solid dispersion of PRX-PEG 4000, prepared with varying concentrations of poloxamer 188, resulted in a yellowish-white solid. The solid dispersion was then weighed, and the percentage yield was calculated to assess the material loss during the preparation process. The percentage yields for the formulations with P188 concentrations of 0%, 1%, 3%, and 5% were 98.18%, 97.40%, 98.26%, and 98.58%, respectively. Notably, the highest yield was observed at the 5% P188 concentration. These results suggested that variations in P188 concentration did not significantly affect the percentage yield of the solid dispersion.
Solubility Study
The solubility study was performed to evaluate the dissolution capacity of the drug in aqueous media, using distilled water as the solvent. The process was monitored over 24 h, with samples collected at predetermined intervals. The resulting solubility profiles for each solid dispersion formula are shown in Figure 1. From these profiles, several key parameters were identified to guide the selection of the optimal solid dispersion formulation. These parameters include saturation solubility (S₀), maximum concentration (Cmax), area under the curve (AUC), dissolution rate, precipitation rate, and the Cmax: S₀ ratio. The average values for each parameter across the different formulas are summarized in Table 1.
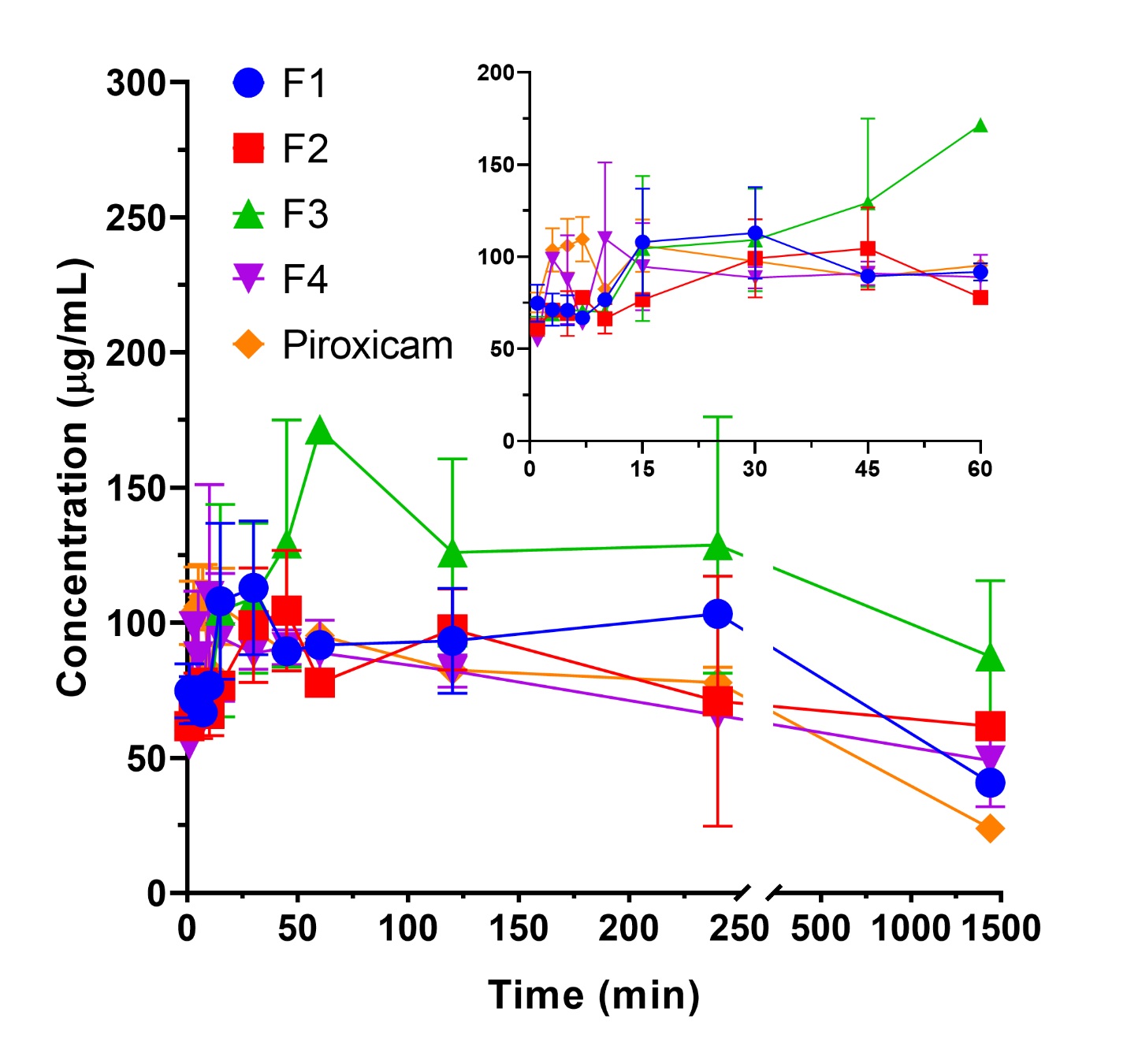
Saturation solubility was determined at the 1440-minute time point, in which it is assumed that the drug has reached saturation conditions (33). Overall, the saturation solubility of all solid dispersion formulations was higher than that of the pure PRX formulation, with increases of 1.7, 1.06, 4.65, and 2.76 times for Formula 1, Formula 2, Formula 3, and Formula 4, respectively. The saturated solubility, ranked from highest to lowest, was as follows: Formula 3 > Formula 4 > Formula 1 > Formula 2, with values ranging from 110.83 µg/mL (Formula 3) to 25.18 µg/mL (Formula 2). Notably, Formula 3 exhibited the highest solubility, which could be attributed to the optimal combination of excipients used in the solid dispersion formulation. These results suggest that the solid dispersion technique, particularly with the formulation used in Formula 3, has a significant potential to enhance the solubility of PRX compared to the pure drug.
The maximum concentration (Cmax) represents the peak concentration of the drug dissolved in the medium. Higher Cmax values are indicative of better performance (Sun and Lee, 2015). The formulations ranked from highest to lowest Cmax are Formula 1, Formula 3, Formula 2, and Formula 4, with the highest Cmax of 97.421 µg/mL (Formula 1) and the lowest of 86.260 µg/mL (Formula 4). The AUC, calculated from the area under the dissolution curve from the 1st minute to the 1440th minute, is an important indicator of bioavailability. A higher AUC suggests improved drug absorption and overall efficacy (34). The AUC values, ranked from highest to lowest, are Formula 3, Formula 2, Formula 1, and Formula 4, with the highest AUC of 112, 002 µg. min/mL (Formula 3), indicating more sustained solubilization and the lowest of 93, 811.1 µg. min/mL (Formula 4). This finding suggests that moderate P188 concentrations (3%) facilitate a balance between dissolution enhancement and matrix stability, resulting in superior solubilization performance.
The dissolution rate was calculated as the slope of the concentration-time curve obtained from linear regression, while the precipitation rate was determined from the slope of the decreasing concentration over time (35-37). The dissolution rates, from highest to lowest, are Formula 4, Formula 1, Formula 3, and Formula 2, with the highest dissolution rate of 1.066 µg/min (Formula 4) and the lowest of 0.608 µg/min (Formula 2). Conversely, the precipitation rates, ranked from highest to lowest, were Formula 1, Formula 4, Formula 2, and Formula 3, showing that Formula 3 exhibited the slowest precipitation rate and therefore the most stable supersaturated state.
The final parameter, the Cmax: S₀ ratio, indicates the degree of supersaturation in the formulation (38). The Cmax: S₀ ratios, ranked from highest to lowest, are Formula 1, Formula 4, Formula 2, and Formula 3, with the highest ratio being 2.912 (Formula 1) and the lowest 1.571 (Formula 3). Despite its lower Cmax: S₀ ratio, Formula 3 demonstrated a more favorable balance of high solubility, reduced precipitation, and stable molecular dispersion, supporting its selection as the optimal formulation.
These findings align with previous studies showing that the inclusion of poloxamers at moderate levels can improve solubility and dissolution through enhanced wettability and micellar solubilization (21, 23). However, excessive poloxamer content may lead to gel layer formation during dissolution, reducing effective drug release (39). The improved solubility observed for Formula 3 may therefore be attributed to optimized polymer–surfactant interactions, which prevent recrystallization while maintaining adequate matrix porosity for water penetration.
In summary, all solid dispersion formulations demonstrated an enhancement in solubility relative to the pure PRX formulation. Formula 3, which contains 3% poloxamer 188, was identified as the optimal choice, as it exhibited the best performance in four of the six evaluated parameters: saturation solubility, Cmax: S₀ ratio, precipitation rate, and AUC (Table 1). This improvement is likely due to the surface-active and wetting properties of poloxamer 188, which enhance drug solubility by facilitating rapid hydration upon contact with the dissolution medium (21). However, higher concentrations of poloxamer 188 may impede the dissolution rate due to the gelation properties of the surfactant (40). Additionally, the presence of surfactants in the solid dispersion system may prevent drug recrystallization, further enhancing dissolution rates (20). These results support that the controlled incorporation of amphiphilic polymers, such as poloxamer 188, can effectively improve solubility and maintain supersaturation stability, consistent with previous reports on solid dispersion systems for poorly soluble drugs.
| Formula | S0 (µg/mL) | Cmax (µg/mL) | Cmax/S0 ratio | Dissolution Rate (µg/min) | Precipitation Rate (µg/min) | AUC (µg. min/mL) |
|---|---|---|---|---|---|---|
| F1 | 40.81 ± 6.12 | 97.42 ± 0.36 | 2.91 ± 0.79 | 1.07 ± 0.61 | 0.31 ± 0.05 | 96171.2 ± 20174.0 |
| F2 | 25.18 ± 0.82 | 88.46 ± 13.04 | 2.12 ± 1.56 | 0.61 ± 0.39 | 0.06 ± 0.03 | 109555.7 ± 31818.3 |
| F3 | 110.83 ± 38.47 | 93.60 ± 9.10 | 1.57 ± 0.50* | 1.00 ± 0.06 | 0.04 ± 0.00 | 112002.3 ± 27374.8 |
| F4 | 65.69 ± 7.37 | 86.26 ± 9.54 | 2.13 ± 1.15 | 1.07 ± 0.76 | 0.10 ± 0.05 | 93811.1 ± 26813.1 |
| PRX | 23.83 ± 1.77 | 95.72 ± 3.93 | 4.59 ± 1.22 | 0.89 ± 0.04 | 0.09 ± 0.05 | 81755.1 ± 6518.5 |
| Note: *One-way ANOVA revealed a statistically significant difference compared with the PRX group (p < 0.05) | ||||||
Fourier Transform Infrared Spectroscopy (FTIR)
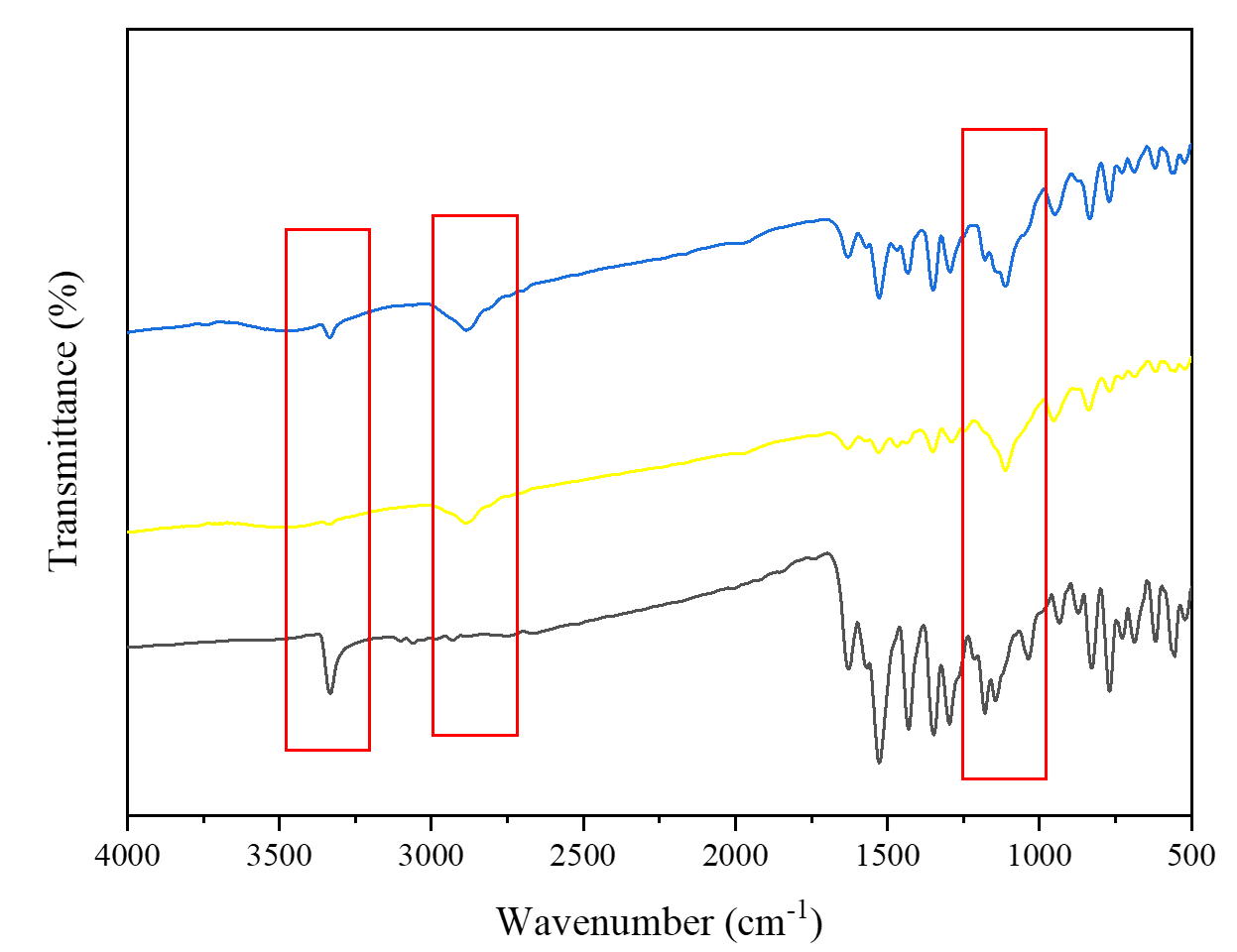
The FTIR analysis was conducted to assess the presence of intermolecular interactions between the drug and one or both carriers, which can be indicated by shifts in peaks or the emergence of new peaks. The FTIR spectra revealed differences between the PRX solid dispersions, pure PRX, and physical mixtures (Figure 2). Specifically, in the 3500–3250 cm⁻¹ range, which corresponds to the -NH group, a distinct peak was observed in the spectra of pure PRX and physical mixtures. In contrast, this peak was absent in the spectra of the PRX solid dispersions. These findings suggest the formation of molecular interactions, likely through hydrogen bonding between the drug and the lone electron pairs of the oxygen atoms in PEG 4000 or poloxamer 188. Such interactions may inhibit the crystallization of the drug, leading to enhanced solubility and dissolution rate (41-43). These findings are consistent with previous reports where PEG-poloxamer-based solid dispersions facilitated drug-polymer hydrogen bonding, improving molecular dispersion and reducing crystallinity (23, 44). The formation of these intermolecular interactions likely plays a central role in stabilizing PRX in an amorphous or partially amorphous state within the polymeric matrix.
Differential Scanning Calorimetry (DSC)
The DSC analysis was conducted to evaluate the formation of the solid dispersion, based on differences in the thermal profiles of the individual components. The thermogram results are shown in Figure 3. Pure PRX exhibited a sharp endothermic peak at 203.12°C, corresponding to its melting point, indicating its crystalline nature. PEG 4000 and poloxamer 188 showed endothermic peaks at 67.14°C and 60.39°C, respectively. In contrast, the DSC thermogram of the PRX solid dispersion displayed a single broad endothermic peak around 64.47 °C, corresponding to the melting range of the carriers, with the characteristic PRX melting peak completely absent. This disappearance, along with the reduced enthalpy change (ΔH = –48.97 J/g), confirms the transition of PRX from a crystalline to an amorphous or molecularly dispersed form within the carrier matrix. These results indicate the successful formation of a solid dispersion, with no crystalline PRX present, and the solid dispersion exhibits thermal properties similar to those of the carrier (14, 45).
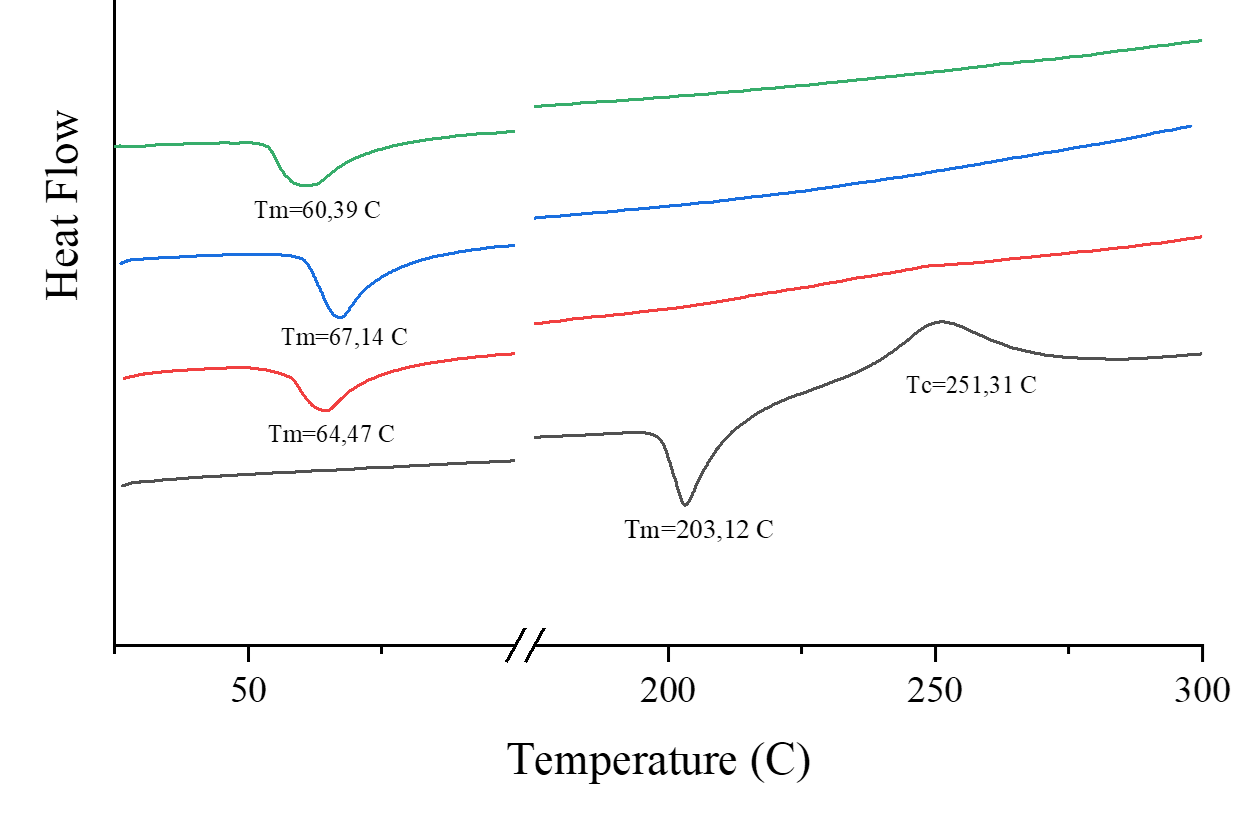
The consistent evidence of molecular interaction, peak shifts, and loss of crystalline thermal signatures collectively supports the successful formation of the PRX-PEG4000-P188 solid dispersion system. These results align with previous work but extend the understanding of optimal carrier composition for PRX, demonstrating that moderate inclusion of poloxamer 188 (3%) achieves a favorable balance between solubilization and matrix stability.
Evaluation of Physicochemical Properties of PRX Solid Dispersion Capsules
Angle of Repose
The angle of repose study was conducted to assess the flow properties of the preparation. A smaller angle of repose indicates better flow properties (46). The results revealed that the angle of repose for pure PRX capsules and PRX solid dispersions was 27.4 ± 0.19 and 25.6 ± 0.10°, respectively. Both formulations exhibited excellent flow properties, as their angle of repose values fall within the range of 25–30°, which is generally considered ideal for optimal flow (30). This can be attributed to the presence of additional lubricants, such as magnesium stearate, in the capsule formulation, which is known to enhance flowability by reducing friction between particles (47).
Furthermore, the slight reduction in the angle of repose for the PRX solid dispersions compared to the pure PRX capsules may be indicative of improved particle size distribution and uniformity in the solid dispersion system. The presence of PEG 4000 and P188 in the solid dispersion could also contribute to better flow properties by acting as surfactants that reduce interparticle adhesion and enhance the powder’s flowability.
Uniformity of Weight
The weight uniformity study is crucial for ensuring the consistency and reliability of capsule content across a batch. In this study, the average capsule weight for pure PRX was 264.1 mg, with a relatively low deviation of 0.92%. For the solid dispersion capsules, the average weight was slightly higher at 265.9 mg, with a slightly greater deviation of 1.06%. Despite this slight difference in the average weights and deviations, both formulations met the standard requirements for weight uniformity, demonstrating consistent and reproducible results (48). These findings suggest that the encapsulation process used for both pure PRX and solid dispersion formulations was well-controlled, ensuring uniformity across capsules.
Disintegration Time
The disintegration time study is designed to evaluate how quickly a capsule breaks down under specific conditions, which is critical for ensuring that the drug is released efficiently (32). In this study, the average disintegration time for the pure PRX capsules was 1 minute and 21 s, while the PRX solid dispersion capsules took slightly longer at 1 minute and 26 s. Both formulations exhibited favorable disintegration times, well within the expected range for effective release. This rapid disintegration can be attributed to the inclusion of sodium starch glycolate (SSG) as a disintegrant in the capsule formulation, which facilitates faster breakdown and drug release (49).
Validation of PRX Analysis Method
The validation of the analytical method was conducted through precision, accuracy, and placebo interference testing. Precision was assessed to determine the repeatability and variation of the analytical method. This was done by analyzing samples at three different concentrations, each with three replications. Accuracy, on the other hand, reflects how close the analytical value is to the actual or expected value. The method is considered precise if the relative standard deviation (RSD) is < 2%, and accurate if the percentage recovery is within the range of 98-102% (50). In this study, the RSD value was found to be 0.94%, and the percentage recovery was 98.89%, both of which meet the required criteria for precision and accuracy.
Placebo interference testing was performed to assess the potential impact of other materials on the analysis. In this case, the placebo interference was found to be 1.299%, which is well below the accepted threshold of 2% (32). This suggests that the additional materials used did not interfere with the analysis results, confirming the method's robustness. In conclusion, these results demonstrate that the analytical method meets all validation requirements, ensuring its reliability and accuracy for further use.
Dissolution Study
The dissolution study was conducted to evaluate the impact of drug modification through a solid dispersion system on the bioavailability of PRX, comparing it to the dissolution profile of pure PRX capsules in vitro. According to the dissolution test results, the selected solid dispersion formula demonstrated a 77.58% drug dissolution at the 45th minute, whereas the pure PRX capsules showed only 55.36% dissolution (Figure 4). This represents a 22.22% increase in drug dissolution for the solid dispersion formulation. To further analyze the dissolution profiles, the similarity factor (F2) was calculated, yielding a value of 36.01. An F2 value below 50 indicates that the dissolution profiles of the solid dispersion capsules and the pure PRX capsules were dissimilar. Therefore, it can be concluded that the solid dispersion system significantly enhances the dissolution rate of PRX.
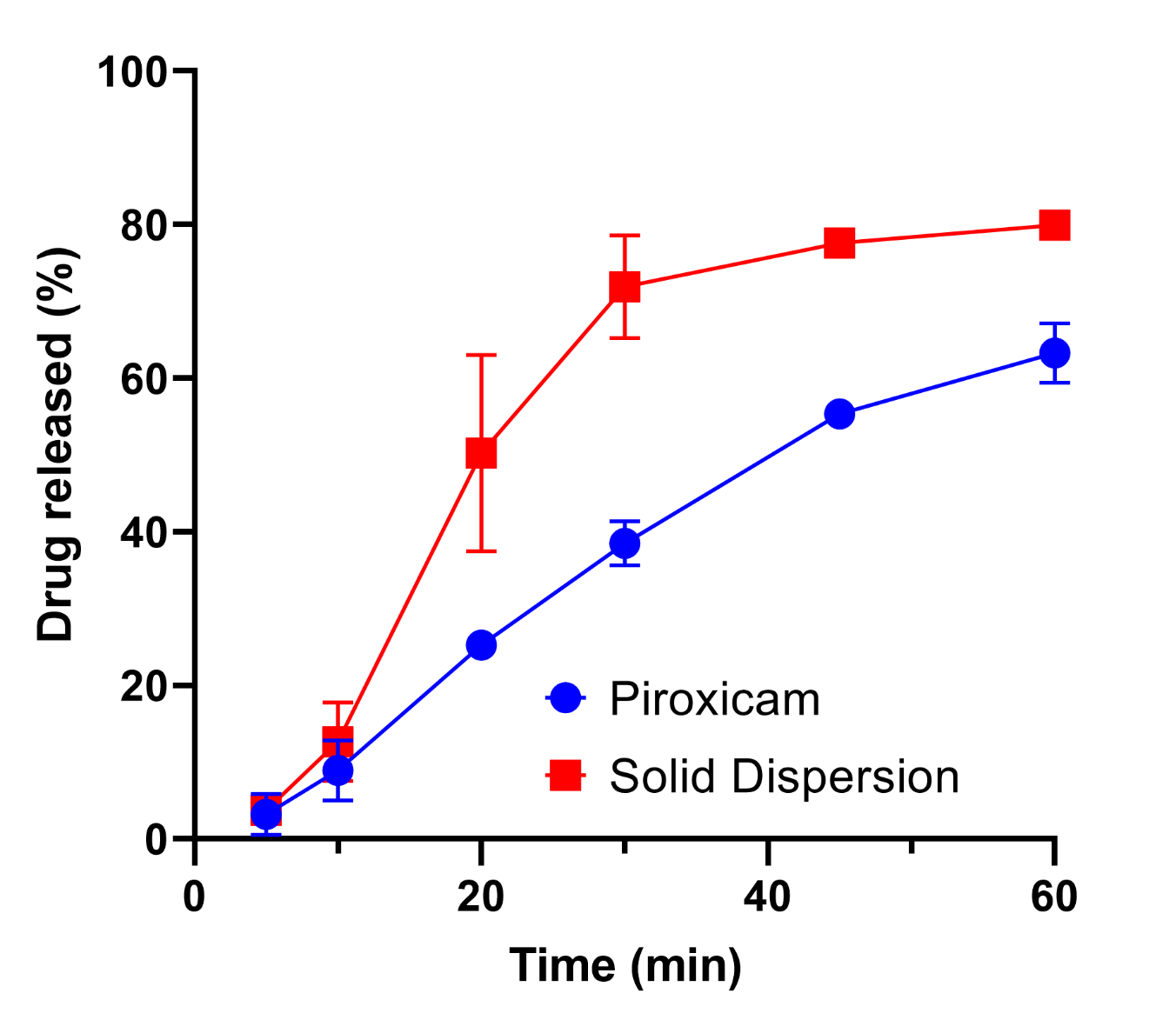
This improvement in dissolution can be attributed to several factors. The solid dispersion system promotes drug dissolution through both diffusion and the influence of surfactants, which reduces the interfacial tension between the drug and the dissolution medium. Additionally, the molecular interactions between the drug and the carrier further contribute to this enhanced dissolution (43, 51, 52). The extent of improvement achieved in this study is consistent with previously reported PRX solid dispersion studies using single hydrophilic carriers such as PEG or PVP, which demonstrated dissolution enhancements (8, 19, 23). However, our PEG–poloxamer binary system offers a simpler and solvent-free approach, with improved formulation stability and scalability compared to nanosuspension or cyclodextrin-based systems (10, 22, 53). This comparative advantage highlights the novelty of the current work, emphasizing the potential of moderate poloxamer incorporation to strike a balance between solubility enhancement and formulation practicality.
Nevertheless, several limitations of the present study should be acknowledged. First, the dissolution tests were conducted only in artificial gastric fluid. Future studies employing biorelevant media, such as simulated intestinal fluid (SIF), would provide more predictive insights into in vivo performance. Second, stability studies were not performed; therefore, the long-term physical stability of the amorphous solid dispersion remains to be confirmed. Third, only a limited range of poloxamer concentrations (0–5%) was evaluated. Exploring a broader concentration range or alternative surfactant types may yield further optimization of dissolution performance. Overall, despite these limitations, the findings clearly demonstrate that incorporating a small amount of poloxamer 188 (3%) into a PEG 4000-based solid dispersion significantly enhances PRX solubility and dissolution. This formulation strategy thus represents a practical and scalable approach to improve the oral bioavailability of poorly soluble NSAIDs such as piroxicam.
Conclusion
In conclusion, the PRX-PEG4000-P188 solid dispersion system, prepared using the melting method, significantly enhances the solubility of PRX. Among the formulations, the solid dispersion containing 3% poloxamer 188 showed the most favorable solubility results. Moreover, the PRX solid dispersion capsules exhibited an improved dissolution profile compared to pure PRX capsules, with a notable 22.22% increase in dissolution rate. These findings indicate that PRX-PEG4000-P188 solid dispersion systems are a promising approach for improving the solubility and dissolution rate of PRX. Nevertheless, further in vivo studies are needed to evaluate their potential impact on therapeutic efficacy.
Abbreviations
BCS = Biopharmaceutical Classification System; NSAID = Non-Steroidal Anti-Inflammatory Drug; PRX = Piroxicam; PEG= Polyethylene Glycol; DSC = Differential Scanning Calorimetry; FTIR = Fourier Transform Infrared Spectroscopy; HCl = Hydrochloric Acid; S0 = saturation solubility; Cmax = Maximum Concentration; AUC = Area Under the Curve; RSD = Relative Standard Deviation; F2 = Similarity Factor.
Declarations
Acknowledgment
The authors would like to express their gratitude to Universitas Sebelas Maret for the partial financial support provided for this research.
Conflict of Interest
The authors declare no conflicting interest.
Data Availability
The unpublished data is available upon request to the corresponding author.
Funding Information
This research was conducted partially funded by Universitas Sebelas Maret, under grant number 371/UN27.22/PT.01.03/2025.
References
- Lindenberg M, Kopp S, Dressman JB. Classification of orally administered drugs on the World Health Organization Model list of Essential Medicines according to the biopharmaceutics classification system. European Journal of Pharmaceutics and Biopharmaceutics. 2004 Sep;58(2):265–78.
- Ting JM, Porter WW, Mecca JM, Bates FS, Reineke TM. Advances in Polymer Design for Enhancing Oral Drug Solubility and Delivery. Bioconjug Chem. 2018 Apr 18;29(4):939–52.
- Chantasart D, Tocanitchart P, Wongrakpanich A, Teeranachaideekul V, Junyaprasert VB. Fabrication and evaluation of Eudragit® polymeric films for transdermal delivery of piroxicam. Pharm Dev Technol. 2018 Sep 14;23(8):771–9.
- Saganuwan SA. Physicochemical and structure-activity properties of piroxicam—a mini review. Comp Clin Path. 2016 Sep 1;25(5):941–5.
- Tantishaiyakul V, Permkam P, Suknuntha K. Use of drifts and PLS for the determination of polymorphs of piroxicam alone and in combination with pharmaceutical excipients: a technical note. AAPS PharmSciTech. 2008 Mar;9(1):95–9.
- Bhalani D V., Nutan B, Kumar A, Singh Chandel AK. Bioavailability Enhancement Techniques for Poorly Aqueous Soluble Drugs and Therapeutics. Biomedicines. 2022 Sep 1;10(9):2055.
- Swidan SA, Ghonaim HM, Ghorab MM, Samy AM. Design, Formulation and Evaluation of Piroxicam Capsules Prepared by Solid Dispersion Technique. J Pharm Res Int. 2013 Mar 3;3(1):108–34.
- Pan RN, Chen JH, Chen RRL. Enhancement of dissolution and bioavailability of piroxicam in solid dispersion systems. Drug Dev Ind Pharm. 2000;26(9):989–94.
- Alhamhoom Y, Honmane SM, Hani U, Osmani RAM, Kandasamy G, Vasudevan R, et al. Study of Formulation and Process Variables for Optimization of Piroxicam Nanosuspension Using 32 Factorial Design to Improve Solubility and In Vitro Bioavailability. Polymers 2023, Vol 15, Page 483. 2023 Jan 17;15(3):483.
- Aksoy OA, Zanbak Çotaoğlu M, Fatsa T, Topal GR, Eşim Ö, Göksel BA, et al. Preparation of Piroxicam nanosuspensions by high pressure homogenization and evaluation of improved bioavailability. Drug Dev Ind Pharm. 2023;49(12):715–22.
- Jug M, Bećirević-Laćan M. Multicomponent complexes of piroxicam with cyclodextrins and hydroxypropyl methylcellulose. Drug Dev Ind Pharm. 2004;30(10):1051–60.
- Xiliang G, Yu Y, Guoyan Z, Guomei Z, Jianbin C, Shaomin S. Study on inclusion interaction of piroxicam with β-cyclodextrin derivatives. Spectrochim Acta A Mol Biomol Spectrosc. 2003 Dec 1;59(14):3379–86.
- Sareen S, Mathew G, Joseph L. Improvement in solubility of poor water-soluble drugs by solid dispersion. Int J Pharm Investig. 2012;2(1):12.
- Sohn JS, Kim EJ, Park JW, Choi JS. Piroxicam ternary solid dispersion system for improvement of dissolution (%) and in vitro anti-inflammation effects. Materials Science and Engineering: B. 2020 Nov 1;261:114651.
- Williams HD, Trevaskis NL, Charman SA, Shanker RM, Charman WN, Pouton CW, et al. Strategies to address low drug solubility in discovery and development. Pharmacol Rev. 2013;65(1):315–499.
- Soyata A, Kenti K, Sutoro M, Sagita N. Impact of Preparation Method in Co-Amorphous System. Sciences of Pharmacy. 2022 Jun 28;1(1):47–55.
- Cid AG, Simonazzi A, Palma SD, Bermúdez JM. Solid dispersion technology as a strategy to improve the bioavailability of poorly soluble drugs. Ther Deliv. 2019;10(6):363–82.
- Li J, Miao X, Chen T, Ouyang D, Zheng Y. Preparation and characterization of pelletized solid dispersion of resveratrol with mesoporous silica microparticles to improve dissolution by fluid-bed coating techniques. Asian J Pharm Sci. 2016 Aug 1;11(4):528–35.
- Barzegar-Jalali M, Ghanbarzadeh S, Adibkia K, Valizadeh H, Bibak S, Mohammadi G, et al. Development and characterization of solid dispersion of piroxicam for improvement of dissolution rate using hydrophilic carriers. Bioimpacts. 2014;4(3):141.
- Chaudhari SP, Dugar RP. Application of surfactants in solid dispersion technology for improving solubility of poorly water soluble drugs. J Drug Deliv Sci Technol. 2017 Oct 1;41:68–77.
- Tambe A, Pandita N. Enhanced solubility and drug release profile of boswellic acid using a poloxamer-based solid dispersion technique. J Drug Deliv Sci Technol. 2018 Apr 1;44:172–80.
- Vo CLN, Park C, Lee BJ. Current trends and future perspectives of solid dispersions containing poorly water-soluble drugs. Eur J Pharm Biopharm. 2013;85(3 Pt B):799–813.
- El-Badry M, Fathy M. Properties of solid dispersion of piroxicam in Pluronic F-98. J Drug Deliv Sci Technol. 2004 Jan 1;14(3):199–205.
- Bethune SJ, Schultheiss N, Henck JO. Improving the poor aqueous solubility of nutraceutical compound pterostilbene through cocrystal formation. Cryst Growth Des. 2011 Jul 6;11(7):2817–23.
- Alotaibi BS, Khan MA, Ullah K, Yasin H, Mannan A, Khan SA, et al. Formulation and characterization of glipizide solid dosage form with enhanced solubility. PLoS One. 2024 Feb 1;19(2).
- Verhoeven E, De Beer TRM, Van den Mooter G, Remon JP, Vervaet C. Influence of formulation and process parameters on the release characteristics of ethylcellulose sustained-release mini-matrices produced by hot-melt extrusion. European Journal of Pharmaceutics and Biopharmaceutics. 2008 May 1;69(1):312–9.
- Paudwal G, Dolkar R, Perveen S, Sharma R, Singh PP, Gupta PN. Third Generation Solid Dispersion-Based Formulation of Novel Anti-Tubercular Agent Exhibited Improvement in Solubility, Dissolution and Biological Activity. AAPS Journal. 2024 Jun 1;26(3):1–14.
- Baghel S, Cathcart H, O’Reilly NJ. Polymeric Amorphous Solid Dispersions: A Review of Amorphization, Crystallization, Stabilization, Solid-State Characterization, and Aqueous Solubilization of Biopharmaceutical Classification System Class II Drugs. J Pharm Sci. 2016 Sep 1;105(9):2527–44.
- Wu KW, Sweeney C, Dudhipala N, Lakhani P, Chaurasiya ND, Tekwani BL, et al. Primaquine loaded solid lipid nanoparticles (SLN), nanostructured lipid carriers (NLC), and nanoemulsion (NE): effect of lipid matrix and surfactant on drug entrapment, in vitro release, and ex vivo hemolysis. AAPS PharmSciTech. 2021 Oct 1;22(7):240.
- Beakawi Al-Hashemi HM, Baghabra Al-Amoudi OS. A review on the angle of repose of granular materials. Powder Technol. 2018 May 1;330:397–417.
- Mohylyuk V, Bandere D. High-Speed Tableting of High Drug-Loaded Tablets Prepared from Fluid-Bed Granulated Isoniazid. Pharmaceutics 2023, Vol 15, Page 1236. 2023 Apr 13;15(4):1236.
- Kemenkes RI. Farmakope Indonesia. VI. Jakarta; 2020.
- Panzade P, Shendarkar G, Shaikh S, Rathi PB. Pharmaceutical Cocrystal of Piroxicam: Design, Formulation and Evaluation. Adv Pharm Bull. 2017;7(3):399.
- Sun DD, Lee PI. Evolution of supersaturation of amorphous pharmaceuticals: Nonlinear rate of supersaturation generation regulated by matrix diffusion. Mol Pharm. 2015 Mar 16;12(4):1203–15.
- Anhalt K, Geissler S, Harms M, Weigandt M, Fricker G. Development of a new method to assess nanocrystal dissolution based on light scattering. Pharm Res. 2012 Oct;29(10):2887–901.
- Chauhan H, Kuldipkumar A, Barder T, Medek A, Gu CH, Atef E. Correlation of inhibitory effects of polymers on indomethacin precipitation in solution and amorphous solid crystallization based on molecular interaction. Pharm Res. 2014 Feb;31(2):500–15.
- Warren DB, Benameur H, Porter CJH, Pouton CW. Using polymeric precipitation inhibitors to improve the absorption of poorly water-soluble drugs: A mechanistic basis for utility. J Drug Target. 2010 Dec;18(10):704–31.
- Sun DD, Wen H, Taylor LS. Non-Sink Dissolution Conditions for Predicting Product Quality and In Vivo Performance of Supersaturating Drug Delivery Systems. J Pharm Sci. 2016 Sep 1;105(9):2477–88.
- Abdeltawab H, Svirskis D, Sharma M. Formulation strategies to modulate drug release from poloxamer based in situ gelling systems. Expert Opin Drug Deliv. 2020 Apr 2;17(4):495–509.
- Medarević DP, Kachrimanis K, Mitrić M, Djuriš J, Djurić Z, Ibrić S. Dissolution rate enhancement and physicochemical characterization of carbamazepine-poloxamer solid dispersions. Pharm Dev Technol. 2016 Apr 2;21(3):268–76.
- Choi JS, Park JW, Park JS. Design of Coenzyme Q10 solid dispersion for improved solubilization and stability. Int J Pharm. 2019 Dec 15;572.
- Koh P, Chuah J, Talekar M, Gorajana A, Garg S. Formulation Development and Dissolution Rate Enhancement of Efavirenz by Solid Dispersion Systems. Indian J Pharm Sci. 2013 May;75(3):291.
- Schittny A, Huwyler J, Puchkov M. Mechanisms of increased bioavailability through amorphous solid dispersions: a review. Drug Deliv. 2020 Jan 1;27(1):110–27.
- Zhang J, Guo M, Luo M, Cai T. Advances in the development of amorphous solid dispersions: The role of polymeric carriers. Asian J Pharm Sci. 2023 Jul 1;18(4):100834.
- Sankari T, Al-Hariri S. Preparation and characterization of cefuroxime axetil solid dispersions using poloxamer 188. Brazilian Journal of Pharmaceutical Sciences. 2019 Apr 8;54(4):e17644.
- Stranzinger S, Faulhammer E, Scheibelhofer O, Calzolari V, Biserni S, Paudel A, et al. Study of a low-dose capsule filling process by dynamic and static tests for advanced process understanding. Int J Pharm. 2018 Apr 5;540(1–2):22–30.
- Morin G, Briens L. The effect of lubricants on powder flowability for pharmaceutical application. AAPS PharmSciTech. 2013 Sep;14(3):1158–68.
- Osorio JG, Muzzio FJ. Effects of powder flow properties on capsule filling weight uniformity. Drug Dev Ind Pharm. 2013 Sep;39(9):1464–75.
- Agrawal A, Dudhedia M, Deng W, Shepard K, Zhong L, Povilaitis E, et al. Development of Tablet Formulation of Amorphous Solid Dispersions Prepared by Hot Melt Extrusion Using Quality by Design Approach. AAPS PharmSciTech. 2016 Feb 1;17(1):214–32.
- Ahmad N, Bitar Y, Trefi S. Development and validation of a simple method for the determination of Atorvastatin calcium in pure and pharmaceutical formulations using spectrofluorimetry. Heliyon. 2023 Mar 1;9(3):e13771.
- Tran TTD, Tran PHL. Molecular Interactions in Solid Dispersions of Poorly Water-Soluble Drugs. Pharmaceutics. 2020 Aug 1;12(8):745.
- Hallouard F, Mehenni L, Lahiani-Skiba M, Anouar Y, Skiba M. Solid Dispersions for Oral Administration: An Overview of the Methods for their Preparation. Curr Pharm Des. 2016 Oct 26;22(32):4942–58.
- Scarpignato C. Piroxicam-β-cyclodextrin: a GI safer piroxicam. Curr Med Chem. 2013 Mar 13;20(19):2415–37.