
RESEARCH ARTICLE
Antimalarial Activity of Globimetula oreophila Compounds: In Silico Docking Investigations on Plasmodium falciparum Protease
Academic Editor: Sanchaita Rajkhowa
Sciences of Phytochemistry|Vol. 4, Issue 2, pp. 76-84 (2025)
Views
Downloads
Shares
Received
Mar 15, 2025Revised
Jul 17, 2025Accepted
Aug 15, 2025Published
Sep 3, 2025
Abstract
Malaria remains a major global health challenge due to its high morbidity and mortality, further complicated by growing antimalarial drug resistance. Natural products are being increasingly explored as potential sources of new therapies, with malarial proteases emerging as promising targets due to their essential roles in parasite development, invasion, egress, and hemoglobin degradation. This study evaluates the inhibitory potential of five compounds, quercetrin (DG1), dihydrostilbene (DG2), 4′-methoxy-isoliquiritigenin (DG3), stigmasterol (DG4), and quercetin (DG5), isolated from Globimetula oreophila leaves, using in silico docking against Plasmodium falciparum enzymes. Targets included falcipain-2 and falcipain-3 (cysteine proteases), SERA5 (hemoglobin-processing enzyme), PfDHFR-TS (bifunctional enzyme), and PfCDPK2 (kinase). Docking revealed strong binding affinities through hydrogen bonds, van der Waals forces, and hydrophobic interactions. DG4 showed a high affinity for PfDHFR (-10.3 kcal/mol), comparable to cycloguanil (-10.7 kcal/mol), while DG1 bound firmly to falcipain-2 (-7.9 kcal/mol), falcipain-3 (-7.5 kcal/mol), and PfCDPK2 (-9.0 kcal/mol). Binding to SERA5 ranged from -6.0 to -6.8 kcal/mol. These findings suggest that the tested compounds may act as inhibitors of vital P. falciparum enzymes, holding promise for the development of antimalarial drugs.
Keywords:
Introduction
The pathogen that causes malaria in humans and other mammalian species belongs to the genus Plasmodium (1). Most tropical and subtropical regions, including Asia, America, and Sub-Saharan Africa, including Nigeria, are affected by this disease. Although the Plasmodium genus contains four species known to cause the disease (Plasmodium falciparum, Plasmodium vivax, Plasmodium ovale, and Plasmodium malariae), P. falciparum is the most dangerous and pathogenic (2-7). It affects a variety of hosts and is the cause of the severe form of malaria. The Plasmodium is carried by the infected Anopheles mosquito, which also serves as a vector (7, 8). An infected person may experience fever, neurological symptoms, opisthotonous episodes, seizures, or possibly go into a coma or pass away. In 2022, there were approximately 249 million cases of malaria and 608,000 deaths globally, with sub-Saharan Africa bearing the brunt, accounting for about 94% of these cases and fatalities (1, 7). The 2023 World Malaria Report explores the relationship between climate change and malaria. Variations in temperature, humidity, and precipitation can affect the behavior and survival of Anopheles mosquitoes, which are responsible for spreading malaria. Additionally, extreme weather conditions, such as heatwaves and floods, may directly influence malaria transmission rates and overall disease impact (9). The development of effective malaria control strategies is significantly hindered by the resistance of malaria parasites to many commonly used antimalarial drugs. Currently, the treatment options for chloroquine-resistant P. falciparum infections are limited to artemisinins and artemisinin combination therapies (ACTs). However, decreased sensitivity to ACTs has been observed in some parts of Asia (1, 7). One of the significant challenges in creating new antimalarial candidate drugs is identifying lead compounds with optimal pharmacokinetic properties, including absorption, distribution, metabolism, excretion, and toxicity (ADMET) (10).
Natural product databases offer a practical source for virtual screening against therapeutic targets (11), including parasitic protozoal illnesses (12-14). Natural products have been used as significant leads for drug development: (a) Several natural products are effective drugs despite not falling under the "rule-of-five" (15); (b) despite this, they occupy different regions of biologically relevant chemical space (14), including abundant oxygen-containing functionalities (rarely nitrogen) and high degrees of chirality and complexity (16), (c) they have been evolved to be optimized for activity, including active transport (17), and (d) they can be used as lead structures for semisynthetic modification to increase activity, selectivity, or bioavailability (18). Globimetula oreophila belongs to the Loranthaceae family of parasitic plants, comprising over 75 genera and more than 900 species. It is a member of the hemiparasitic mistletoe family, which is primarily found in tropical Africa, which includes the Central Africa sub-region, Nigeria, Gabon, Congo, and Cameroun (18) Mistletoe uses modified roots to cling to a host plant on a wide range of dicotyledonous trees. Traditional medicine often uses the species to cure a variety of ailments, such as fever, headaches, stomachaches, and diarrhea (19, 20). The G. oreophila plant has previously been subjected to qualitative and quantitative phytochemical screening, which revealed the presence of a variety of secondary metabolites, including alkaloids, flavonoids, carbohydrates, triterpenes, tannins, glycosides, and saponins (20, 21), which have been reported to possess antimalarial activity (22, 23). Additionally, according to Dauda et al. (24), the plant's crude ethanol extract provides a rich source of necessary trace metals in the right amounts, including Zinc (Zn), Cobalt (Co), Copper (Cu), Nickel (Ni), Iron (Fe), and Cadmium (Cd), which supports the plant's therapeutic usefulness in ethnomedicine. According to reports, the Globimetula genus is generally rich in secondary metabolites, and flavonols serve as a marker of the genus's taxonomy (25). There have been prior reports on the antiplasmodial properties of ethanol leaf extract, as well as hexane, chloroform, ethyl acetate, and butanol fractions (7, 20). Previously, we conducted phytochemical investigation studies on the plant and reported the isolation of prenylated quercetin from the ethyl acetate fraction (26). The in-silico analysis of the prenylated quercetin against seven P. falciparum enzymes was also investigated for their antimalarial activity (10). Bio-assay-guided isolation of the hexane, ethyl acetate, and butanol fractions was also carried out to ascertain their antimalarial properties (7). These procedures led to the isolation of five compounds: stigmasterol, quercetin, quercetrin, prenylated dihydrostilbene, and 4′-methoxy-isoliquiritigenin, two of which are novel to this plant's genus and these were characterize and elucidated using spectroscopic techniques such as UV, IR, 1D and 2D NMR, (25, 26). Previously in silico studies, the isolated compounds of G. oreophila showed exceptional binding affinities towards plasmepsin I and II, two main enzymes involved in hemoglobin catabolism throughout intra-erythrocytic development of P. falciparum. These in silico tests identified that molecules such as quercetin and stigmasterol bind strongly with catalytic sites of plasmepsins, suggesting their potential as inhibitors of the proteases (27). Moreover, drug-likeness and toxicity profiling using in silico tools indicated that the isolated compounds of G. oreophila possess excellent oral bioavailability and minimal toxicity levels (27). These findings support their potential to serve as lead compounds for the development of novel antimalarial agents (27). While G. oreophila compounds show activity against plasmepsin I and II (27), their efficacy against other P. falciparum enzymes (e.g., falcipains, SERA5, PfDHFR-TS, and PfCDPK2) remains unexplored, despite these targets’ role in hemoglobin catabolism and parasite egress. This integrated approach aims to identify new inhibitors that would disrupt more than one stage of the parasite's life cycle. Inhibition of multiple enzymes that have multiple functions in multiple stages of the P. falciparum life cycle is a strategic approach to drug resistance. Inhibition of the essential enzymes which function across multiples stages of the parasite’s life cycle-offer a strategic approach to combat drug resistance: falcipains-2/3: critical for hemoglobin degradation, SERA5: mediates merozoite egress; underexplored in drug design, PfDHFR-TS/PfCDPK2: key to nucleotide synthesis and calcium-dependent signaling. In summary, the current research builds upon our previous in silico research, which demonstrated the inhibitory activity of G. oreophila compounds against plasmepsin I and II. We hypothesized that flavonoids (e.g., DG1, DG2, and DG5), prenylated stilbene (DG3), and terpenoid (DG4) from G. oreophila will exhibit strong, multi-target inhibition against cysteine proteases (falcipain-2/3), SERA5, PfDHFR-TS/PfCDPK2 due to structural features (e.g., hydroxylation, prenylation, methoxylation) that align with conserved active sites across these enzymes. This study extends our prior in silico work on plasmepsins (27) to identify novel, multi-target inhibitors that can disrupt critical metabolic processes, thereby providing a multifaceted antimalarial strategy. In the current study, we report the in-silico analysis of G. oreophila secondary metabolites (stigmasterol, quercetin, quercetrin, prenylated dihydrostilbene, and 4′-methoxy-isoliquiritigenin) against P. falciparum enzymes in its life cycle.
Targeting specific enzymes within the malaria parasite and altering its metabolic pathways is a promising strategy for developing new antimalarial drugs. Both synthetic and natural antimalarial agents are designed to disrupt the parasite's unique metabolic processes while sparing the host. During the intra-erythrocytic stage of its lifecycle, the Plasmodium parasite consumes 60–80% of the hemoglobin in red blood cells. It breaks down the hemoglobin to utilize the released amino acids for protein synthesis and energy, while also creating an environment conducive to its growth and replication (10). The degradation of host hemoglobin is facilitated by a series of enzymes, including aspartic proteases (plasmepsins) (10, 27), cysteine proteases (falcipains) (10, 28), and dihydrofolate reductase (10, 29, 30), which release amino acids crucial for the parasite’s nutrition. Additionally, serine proteases (subtilases) play roles in erythrocyte invasion and parasite egress from the host (31, 32). Furthermore, calcium-dependent protein kinase 2, which is critical for calcium signaling throughout various stages of the parasite’s lifecycle, represents a valuable target for both the treatment and prevention of malaria (10).
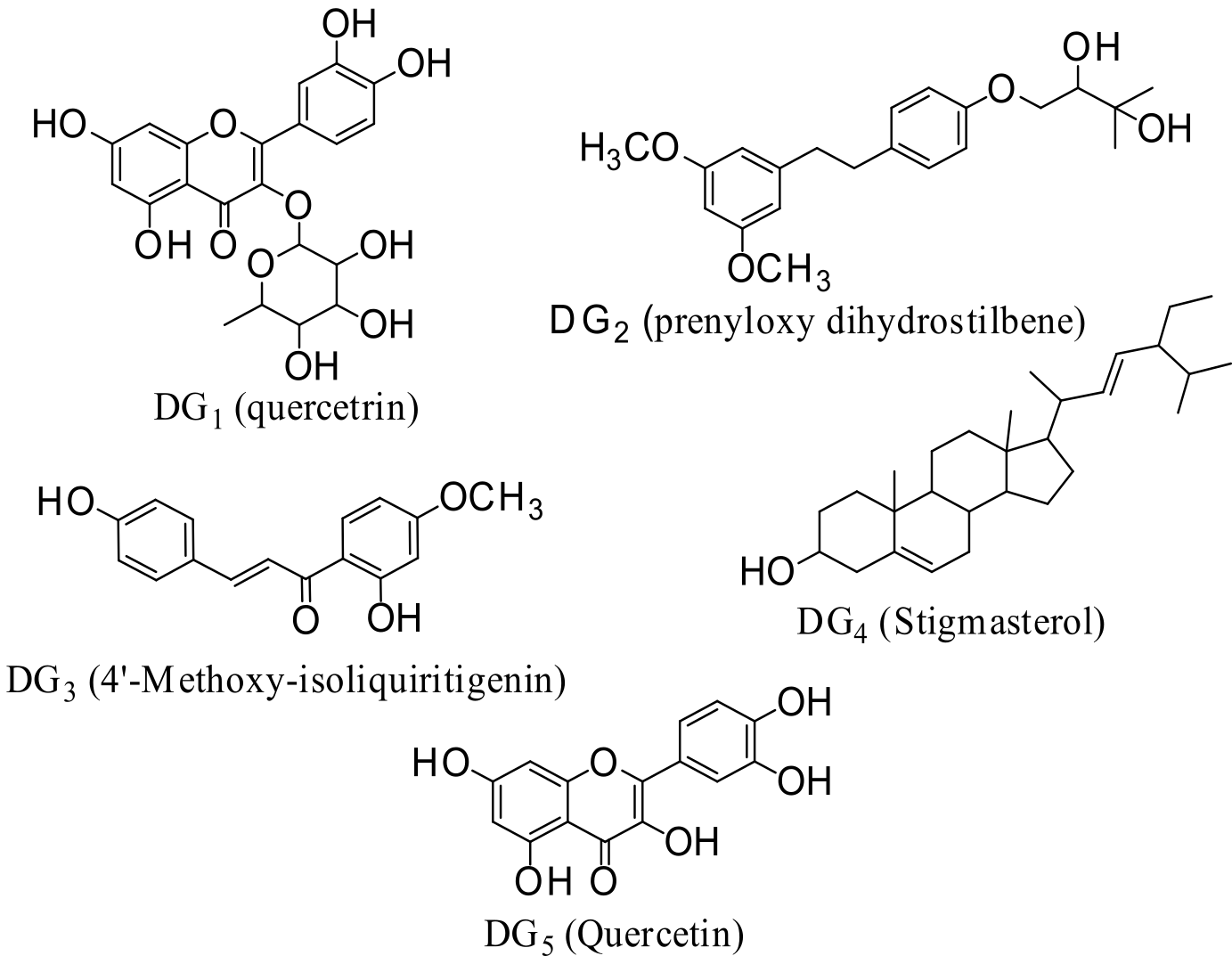
In this study, we utilized a computational approach to qualitatively assess the antimalarial potential of various compounds, optimizing the evaluation process and conserving time. The analysis focused on binding affinities and interactions with the active receptor site of these compounds. The findings suggest that the compounds under investigation could be promising candidates for developing inhibitors targeting proteases and kinases for the treatment of malaria. Specifically, we examined the inhibitory effects of quercetrin (DG1), dihydrostilbene (DG2), 4′-methoxy-isoliquiritigenin (DG3), stigmasterol (DG4), and quercetin (DG5) (Figure 1) isolated from G. oreophila leaves on malaria-related proteins through in-silico docking studies. Our investigation targeted several key P. falciparum enzymes: cysteine proteases (falcipain-2 and -3), serine repeat antigen 5 (SERA5), dihydrofolate reductase-thymidylate synthase (PfDHFR-TS), and calcium-dependent protein kinase 2 (PfCDPK2). The compounds of interest, quercetin, quercetrin, dihydrostilbene, 4′-methoxy-isoliquiritigenin, and stigmasterol, were evaluated for their potential inhibitory activity against these vital enzymes of P. falciparum.
Experimental Section
Software, Hardware, and Databases
AutoDock Vina (33), MGL tools (34), UCSF Chimera (33), ChemDraw Ultra.12, Discovery Studio, Spartan 04, SwissAdme (online server), Mac OSX, Windows (Intel processor, Core i5).
Protein Crystal Structures
High-resolution, non-mutant crystal structure files of the following enzymes from P. falciparum were obtained from RCSB Protein Data Bank (http://www.rcsb.org/pdb accessed on November 17, 2023); Falcipain-2 [FP-2; PDB ID: 6SSZ] (35), Falcipain-3 [FP-3; PDB ID: 3BPM] (36), Serine Repeat Antigen-5 [SERA5; PDB ID: 6X42] (37), P. falciparum Calcium-Dependent Protein Kinase 2 [PfCDPK2; PDB ID: 4MVF] (38), and P. falciparum Dihydrofolate Reductase Thymidylate Synthase [PfDHFR-TS; PDB ID: 4DPD] (39).
In-Silico Antimalarial Studies
A total of five (5) compounds were isolated from the G. oreophila plant: Stigmasterol, quercetin, quercetin-3-rhamnoside, 2’, 4-dihydroxy, 4’-methoxy chalcone, and prenylated dihydrostilbene. Their interaction with a validated crystal structure of some P. falciparum drug targets was studied in silico.
Protein Structure Preparation
As previously stated, the crystal structures were sourced from the Protein Data Bank (PDB). Before the docking procedure, residues within 5.0 Å of the native ligands were analyzed. Using Chimera UCSF, we removed all crystallographic water molecules, ions, and bound ligands from the 3D structures obtained from the PDB (34). The processed receptor structures were then saved as rec.pdb files. AutoDock Tools (33) was utilized to modify the rec.pdb files by incorporating polar hydrogen atoms and Gastegier charges, and these modified files were subsequently saved in pdbqt format.
Ligand Structure Preparation
The 2D structures of the compounds DG1, DG2, DG3, DG4, and DG5 were created using ChemDraw Ultra 12. These 2D structures were then converted to 3D models using Spartan 04. Ligand protonation states were assigned at pH 7.4 (physiological conditions) using ChemAxon’s pKa plugin, ensuring accurate representation of ionizable groups (e.g., hydroxyl, carboxyl) during docking. Geometric optimization of the compounds was performed with the AM1 semi-empirical method in Spartan software, and the optimized 3D structures were saved as mol2 files. AutoDock Tools was employed to add hydrogen atoms and Gastegier charges to these mol2 files, which were then converted and saved in pdbqt format. While Vina’s scoring function neglects entropic penalties and solvent effects, its speed and reproducibility make it suitable for initial screening (27).
Molecular Docking Analysis
Before docking the test compounds, the docking procedure was validated for each protease enzyme by first removing the co-crystallized ligand from the enzyme crystal structure and re-docking it with the specified parameters. The validation was deemed successful if the re-docked ligand conformed closely to the geometrical arrangement of the original co-crystallized ligand in the active site (40). Before conducting molecular docking, the active sites were defined using the coordinates from the crystallographic structures of the enzymes by setting up the grid box (see Table S1). The optimal pose obtained was then used for subsequent analyses. UCSF Chimera was employed for post-docking visualization and for preparing the systems (ligands and receptors) for pre-MD evaluations.
Results and Discussion
Grid Box
The configuration file (config.txt) was customized based on the grid box parameter, leading AutoDock Vina to produce results in pdbqt format. We utilized Chimera's View-Dock function to identify compounds with the best binding energy, optimal geometric conformations, and broad inhibitory activity against all enzymes under investigation. These compounds were then stored in complexes with the reference enzymes. The enzymes and the isolated compounds D1-DG5 (ligands) for each system were carefully prepared using Chimera, adhering to the procedures described by Pettersen and colleagues (34) (Table S2), which provides further details on the grid box parameter.
Validation of Docking Procedures
The docking procedures applied to the seven enzymes were well-validated, as shown in Table S3. All the co-crystallized ligands re-docked on their respective proteins, and are well superimposed on their original Protein Data Bank (PDB) structures.
Binding Energies of Co-crystallized Ligands and Isolated Compounds Against P. falciparum Targets
Binding affinities were interpreted relative to co-crystallized native ligands in each enzyme structure, with values exceeding these benchmarks suggesting more potent potential inhibition. The binding energies of the co-crystallized ligands and the five isolated compounds against protease enzymes are presented in Table 1 and Figure S1.
| Enzyme | Affinity (kcal/mol) | |||||
|---|---|---|---|---|---|---|
| Lig0 | Lig1 | Lig2 | Lig3 | Lig4 | Lig5 | |
| Falcipain-2 | -7.7 | -7.9 | -7.0 | -6.6 | -7.4 | -7.7 |
| Falcipain-3 | -6.5 | -7.5 | -5.8 | -5.6 | -5.4 | -6.5 |
| PfDHFR | -9.7 | -9.5 | -8.2 | -8.2 | -10.3 | -8.5 |
| Serine Repeat Antigen-5 | -4.4 | -6.8 | -6.0 | -6.0 | -6.6 | - 6.7 |
| Calcium Dependent Protein Kinase-2 | -11.7 | -9.0 | -7.5 | -7.5 | -8.6 | -8.3 |
The molecular docking result obtained from this study demonstrated that the test compounds were found to interact with the residues at the active site and other sub-units regulating the specificity of the proteases. The test compounds have a better binding affinity within the binding pockets of falcipain-2, falcipain-3, PfCDPK2, PfDHFR, and SERA5 and which is evident that the binding process where principally favored by hydrogen bond, van der Waals and other hydrophobic interactions. To the binding interactions, the most contributing features of the ligands for receptor interactions are the carbonyl group, hydroxyl group, methyl group, methoxy, oxymethylene group, and the pentacyclic nucleus. Table 1 shows the binding affinities of the test compounds and the standard ligands.
The molecular docking analysis of compound DG1-DG5 (Lig1-Lig5) on falcipain-2 shows binding affinities ranging from -6.6 to -7.8 Kcal/mol, while it was -7.3 Kcal/mol for the native ligand (lig0). The molecular docking results demonstrated that the ligand (Lig1; DG1; -7.8 Kcal/mol) had a better binding affinity within the binding pocket of falcipain-2 than the native ligand (Lig0; native ligand; -7.3 Kcal/mol). From Table 1, the order of increasing binding affinity for the test compounds was -6.6<-6.9<-7.1<7.7<7.8 Kcal/mol (DG3<DG2<DG4<DG5<DG1). A lower free energy indicates a stronger interaction with a receptor. The in-silico study supported the mechanism of action of the isolated compounds to be related to the existence of a 6SSZ receptor that possesses a cysteine protease inhibitor of falcipain-2.
The molecular docking analysis of compound DG1-DG5 (Lig1-Lig5) on falcipain-3 shows docking affinities ranging from -6.6 to -8.2 Kcal/mol, while it was -6.4 Kcal/mol for the native ligand (lig0). The result obtained from the docking analysis demonstrated that the isolated compounds have the least free energy as compared to the native ligand (Lig0), thus resulting in better binding affinity within the binding pocket of falciapin-3 than the native ligand (Lig0). Compounds DG1 and DG5 had the highest binding affinity within the binding pocket of falcipains-3 as compared to other compounds. From Table 1, it was seen that the order of increasing binding energy with the various ligands is -8.2>7.6>6.8>6.6 Kcal/mol (DG1> DG5>DG4>DG3>DG2). The in-silico study suggested that the isolated compounds possess a cysteine protease inhibitor of falcipain-3, thereby disrupting the parasite’s ability to obtain nutrients and survive in the host.
The molecular docking analysis of compound DG1-DG5 (Lig1-Lig5) on PfDHFR-TS shows binding affinities ranging from -8.0 to -10.3 Kcal/mol, while it was -9.6 Kcal/mol for the native ligand (lig0). The molecular docking results demonstrated that the ligands (Lig1- Lig5; DG1-DG5) have better docking affinity within the binding pocket of PfDHFR-TS as compared to the native ligand with binding energy (Lig0; native ligand; -9.6 Kcal/mol). From Table 1, it was seen that the order of increasing binding energy with the various ligands is -10.3>-9.5>8.5>-8.2>-8.0 Kcal/mol (DG4>DG1>DG5>DG3>DG2). The in-silico study predicted that the target site of Lig1-Lig5 is in the gametocytogenesis as a PfDHFR protease inhibitor, which could lead to inhibition of merozoites as a drug target.
The molecular docking analysis of compound DG1-DG5 (Lig1-Lig5) shows docking affinities ranging from -5.4 to -6.6 Kcal/mol, while it was -4.1 Kcal/mol for the native Ligand (Lig0). The result obtained from the docking analysis demonstrated that the isolated compounds have better docking affinity within the binding pocket of SERA5 than the native ligand (Lig0). The lowest binding energy was observed with DG1 compared to other ligands. From Table 1, it was seen that the order of increasing binding energy with the various ligands is -6.6>6.0>-5.9>-5.8>-5.4 Kcal/mol (DG1>DG5>DG3>DG4>DG2). The in-silico study predicted that the target site of Lig1-Lig5 is in the parasitophorous vacuole as a SERA5 protease inhibitor, which could lead to inhibition of merozoites egress as a drug target.
The molecular docking analysis of compound DG1-DG5 (Lig1-Lig5) shows docking affinities ranging from -7.3 to -8.9 Kcal/mol, while it was -11.4 Kcal/mol for the native Ligand (Lig0). The result obtained from the docking analysis demonstrated that the isolated compounds have good docking affinity within the binding pocket of PfCDPK2. But it was seen that the native ligand (Lig0) had the lowest energy as compared to the isolated compounds, resulting in higher binding affinity than the test compounds. From Table 1, it was seen that the order of increasing binding energy with the various ligands is -8.9>8.6>-8.3>-7.4>-7.3 Kcal/mol (DG1>DG4>DG5>DG3>DG2. The in-silico study suggests the mechanism of action of the isolated compounds to be through the existence of a 4MVF receptor that possesses a cyclin-dependent kinase inhibitor of PfCDPK2.
Additionally, the test ligands in all the p. falciparum enzymes had a higher binding affinity than that of the native ligands except in plasmepsin I, II, and PfCDPK2 where the native ligands had a lower binding energy. This implies that the compound can vie with different proteases being investigated for the enzyme's cofactor binding site, ultimately resulting in the suppression of its activity.
Binding Poses and Binding Interactions Analysis of Isolated Compounds Against P. falciparum Enzymes
The binding conformation and interaction of isolated compounds with residues on the active site of the falcipain-2 falcipain-3, CDPK2, SERA5, and PfDHF-TS studied using Chimera (34) and Discovery Studio Suite (www.accelrys.com). Compounds with the lowest binding energy against each enzyme are shown in Figure 2, while other compounds' interactions are shown in (Figure S2-20).
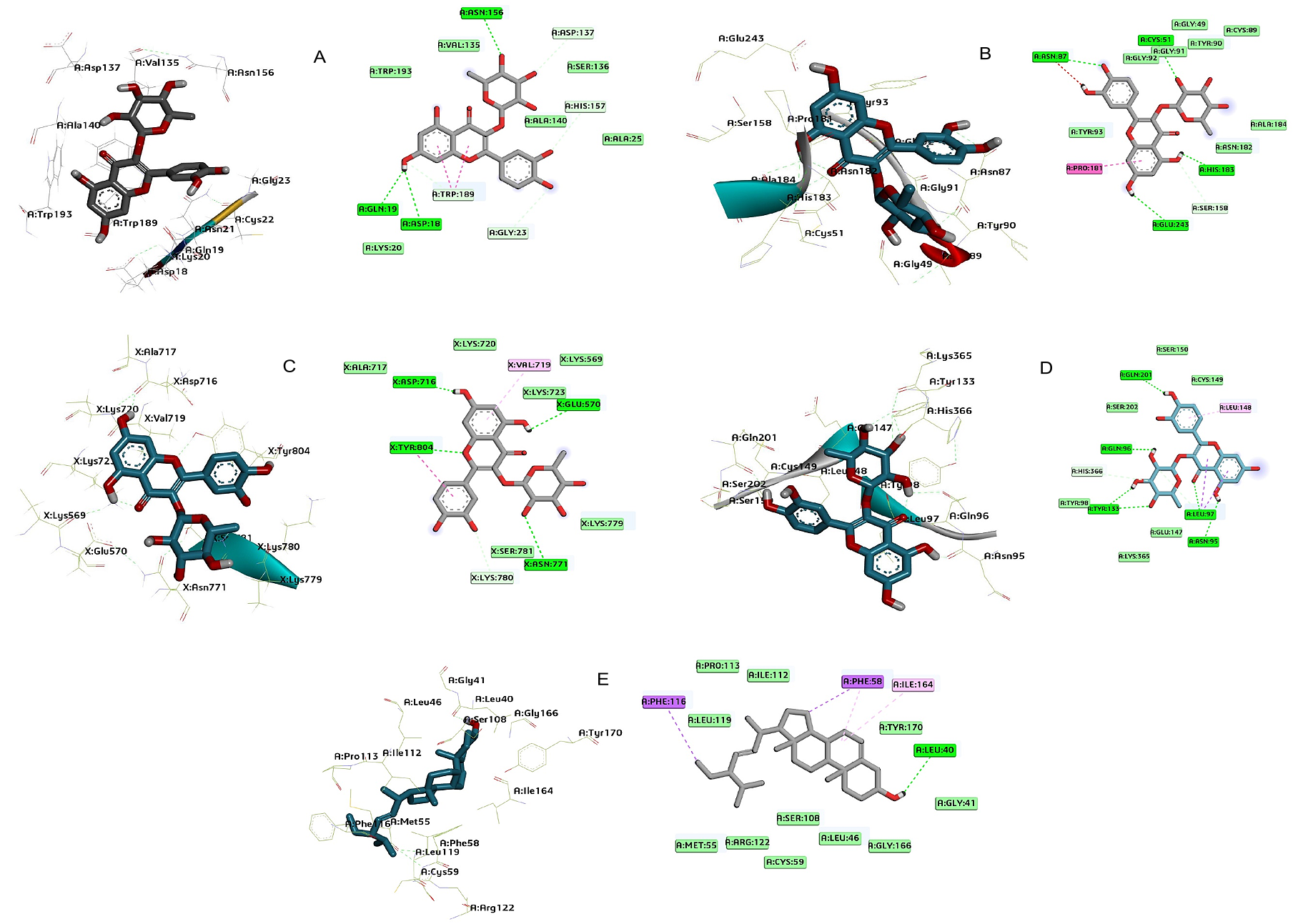
Figures 2-6 depict the 3D and 2D representations illustrating the binding position and interaction analysis of the experimental compound within the active sites of falcipain 2 and 3, P. falciparum dihydrofolate reductase thymidylate synthase (PfDHFR-TS), P. falciparum calcium-dependent protein kinase 2 (PfCDPK2), and serine repeat antigen 5 (SERA5) using Chimera and Discovery Studio Suite. The test compounds were observed to occupy the binding site of the native ligand in the enzymes.
As observed from the molecular interactions of falcipain-2 with quercetrin (DG1), three hydrogen bonds were formed through the ortho-substituted hydroxy group of the aromatic ring and the hydroxy group of the rhamnose sugar moiety in the nucleus of the molecule with the enzyme target, including Asp18, Asn156 and Gln19 (OH) (Figure 2A; Table S4). Hydrophobic interactions were predicted between Falcipain-2 and quercetrin with residues Trp193, Val135, Asp137, Ser136, His157, Ala140, Ala25, Gly23, Trp189a, Lys20, which contributed to the binding affinity of the quercetrin molecule.
Quercetrin (DG1) molecule had four hydrogen bonds, which were formed through a hydroxy group of the ortho-substitution of the aromatic ring and pyranose sugar moiety with falcipain-3 at the active site with the enzymes including Asn87, Glu243, Cys51, and His183. Falcipain-3 has three catalytic triads: Cys51, His183, and Asn213. The amino acids Cys51 and His183 were seen to form hydrogen bond interaction which might be responsible for the inhibition of the parasite growth (Figure 2B; Table S5). Additionally, quercetrin was stabilized through hydrophobic interactions with amino acid residues, including Asn87, Gly92, Gly91, Tyr90, Gly49, Cys89, Ala184, Asn182, Ser158, Pro181, and Tyr93.
From the molecular interactions of quercetrin with serine repeat antigen-5, four hydrogen bonds were formed through the hydroxy group of the aromatic ring, the hydroxy group of the pyrone ring, and that of the sugar moiety hydroxy group with SERA5 at the active site, including Asp716, Glu570, Asn771, and Tyr804 (Figure 2C; Table S6). Additionally, quercetrin was stabilized through hydrophobic interactions with amino acid residues, including Ala717, Lys720, Val719, Lys569, Lys723, Lys779, Ser781, Lys780, and Tyr804.
Quercetrin formed six hydrogen bonds through an ortho-substituted hydroxy group of the phenyl ring, the hydroxy group of the rhamnose sugar moiety, and the carbonyl carbon of the pyrone ring with calcium-dependent protein kinase-2 at the active site, including Gln201 Gln96 Tyr133a (2-bonds; OH) Leu97 Asn95 (Figure 2D; Table S7). Additionally, compound DG1 was stabilized through hydrophobic interactions with amino acid residues such as Ser150, Cys149, Leu148, Leu97a, Glu147, Lys365, Tyr98, His366, and Ser202.
Stigmasterol formed one hydrogen bond with the hydroxyl group of the pentacyclic ring at a beta confirmation with the dihydrofolate reductase-thymidylate synthase, including Leu40 (Figure 2E; Table S8). Additionally, stigmasterol (DG4) was stabilized through hydrophobic interactions with amino acid residues Phe116, Ile112, Leu119, Pro113, Phe58a (2-bonds), Ile164, Tyr170, Gly41, Gly166, Leu46, Ser108, Cys59, Arg122, and Met55.
The discovery of multiple robust bonds formed between the test compounds and the enzymes examined has significantly bolstered the stability of the resulting complexes, thereby elevating the overall binding affinity. This investigation has unveiled compelling evidence showcasing the test compounds' ability to engage actively with key amino acid residues within diverse receptor types. These findings strongly suggest that the compound holds promise in altering the active sites of enzymes associated with P. falciparum, potentially impeding their binding. Such interference could lead to the inhibition of these malaria-related enzymes, disrupting their standard functionality. An intriguing aspect of the compound lies in its consistent structural elements, notably the presence of ortho hydroxyl, carbonyl carbon, methoxy group, and an oxymethylene side chain attached to the molecular nucleus. These features are believed to confer the compound with the capacity to effectively inhibit the enzymes of P. falciparum that were under scrutiny. Noteworthy from the docking simulations are the hydrogen bonds evident in the protein-ligand complexes (as depicted in Figure 2), known to play crucial roles in facilitating protein-ligand interactions. These bonds, along with other interactions like van der Waals forces and electrostatic charges, underscore the high-quality docking and stability observed in this study. Of particular interest is the carbonyl carbon's involvement in hydrogen bonding within the active site, a factor believed to enhance solubility. Furthermore, the presence of various interactions, including van der Waals forces and electrostatic charges, serves as an indicator of robust docking quality and complex stability. The substantial side chain in compound DG2 appears to be more advantageous than smaller side chain groups in ligand-protein interactions, aligning with insights from structure-activity relationships (SARs) (9, 52).
All compounds (DG1-DG5) were previously screened for drug-likeness and ADMET properties (27), demonstrating compliance with Lipinski’s rules (MW<500, LogP≤ 5) and low toxicity risks (ProTox-II).
Conclusion
This study evaluated five metabolites, quercetin (DG1), quercetrin (DG2), dihydrostilbene (DG3), 4′-methoxy-isoliquiritigenin (DG4), and stigmasterol (DG5), from G. oreophila as potential antimalarial agents. Using in silico molecular docking, the compounds were tested against key P. falciparum enzymes: falcipain-2, falcipain-3, SERA5, PfDHFR-TS, and PfCDPK2. Docking results showed strong binding affinities, with DG1 and DG4 emerging as the most promising multi-target candidates (binding energies ≤ –7.5 kcal/mol). Structural features such as ortho-hydroxyl, methoxy, prenyl, and β-hydroxy groups, along with the triterpene skeleton, contributed to stable interactions with catalytic residues (e.g., Tyr77 in falcipain-2, Asp214 in PfDHFR-TS). These findings highlight the compounds’ potential as multi-target inhibitors, though further structure–activity relationship and in vitro studies are needed for confirmation.
Declarations
Acknowledgment
We are thankful to all staff members of the Department of Pharmaceutical and Medicinal Chemistry, Ahmadu Bello University Zaria, Kaduna, Nigeria.
Conflict of Interest
The authors declare no conflicting interest.
Data Availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Funding Information
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Supplemental Material
The following supporting information can be downloaded at: https://etflin.com/file/document/20250315092247666709040.docx. Table S1: Consensus log P values of compound DG1, Table S2: Grid box parameter for the enzymes, Table S3: The crystal structures of enzyme complexes and re-docked ligands super-imposed on the crystal structures for validation, Table S4: Molecular interactions of the amino acid residues of compounds from Azadirachta indica with Falcipain-2 (6SSZ), Table S5: Molecular interactions of the amino acid residues of compounds from Azadirachta indica with Falcipain-3 (3BPM), Table S6: Molecular interactions of the amino acid residues of compounds from Azadirachta indica with Plasmepsin I (3SQ1), Table S7: Molecular interactions of the amino acid residues of compounds from Azadirachta indica with Plasmepsin II (1LF3), Table S9: Molecular interactions of the amino acid residues of compounds from Azadirachta indica with Calcium Dependent Protein Kinase 2 (4MVF), Table S10: Molecular interactions of the amino acid residues of compounds from Azadirachta indica with crystal structures of Plasmodium falciparum vital enzyme DHFR-TS, Figure S1: binding affinities of ligands (DG1-DG5) against Plasmodium falciparum enzymes, Figure S2: 3D molecular pose and 2D interactions of DG2 on the binding cavity of Falcipain-2, Figure S3: 3D molecular pose and 2D interactions of DG3 on the binding cavity of Falcipain-2, Figure S4: 3D molecular pose and 2D interactions of DG4 on the binding cavity of Falcipain-2, Figure S5: 3D molecular pose and 2D interactions of DG5 on the binding cavity of Falcipain-2, Figure S6: 3D molecular pose and 2D interactions of DG2 on the binding cavity of Falcipain-3, Figure S7: 3D molecular pose and 2D interactions of DG3 on the binding cavity of Falcipain-3, Figure S8: 3D molecular pose and 2D interactions of DG4 on the binding cavity of Falcipain-3, Figure 9: 3D molecular pose and 2D interactions of DG1 on the binding cavity of Plasmepsin-I, Figure S10: 3D molecular pose and 2D interactions of DG2 on the binding cavity of Plasmepsin-I, Figure S11: 3D molecular pose and 2D interactions of DG3 on the binding cavity of Plasmepsin-I, Figure S12: 3D molecular pose and 2D interactions of DG5 on the binding cavity of Plasmepsin-I, Figure S13: 3D molecular pose and 2D interactions of DG1 on the binding cavity of Plasmepsin-II, Figure S14: 3D molecular pose and 2D interactions of DG2 on the binding cavity of Plasmepsin-II, Figure S15: 3D molecular pose and 2D interactions of DG3 on the binding cavity of Plasmepsin-II, Figure S16: 3D molecular pose and 2D interactions of DG5 on the binding cavity of Plasmepsin-II, Figure S17: 3D molecular pose and 2D interactions of DG2 on the binding cavity of SERA5, Figure S18: 3D molecular pose and 2D interactions of DG3 on the binding cavity of SERA5, Figure S19: 3D molecular pose and 2D interactions of DG4 on the binding cavity of SERA5, Figure S20: 3D molecular pose and 2D interactions of DG5 on the binding cavity of SERA5, Figure S21: 3D molecular pose and 2D interactions of DG2 on the binding cavity of PfCDPK2, Figure S22: 3D molecular pose and 2D interactions of DG3 on the binding cavity of PfCDPK2, Figure S23: 3D molecular pose and 2D interactions of DG4 on the binding cavity of PfCDPK2, Figure S24: 3D molecular pose and 2D interactions of DG5 on the binding cavity of PfCDPK2, Figure S25: 3D molecular pose and 2D interactions of DG1 on the binding cavity of PfDHFR, Figure S26: 3D molecular pose and 2D interactions of DG2 on the binding cavity of PfDHFR, FigureS27: 3D molecular pose and 2D interactions of DG3 on the binding cavity of PfDHFR, Figure S28: 3D molecular pose and 2D interactions of DG5 on the binding cavity of PfDHFR.
References
- Perlmann P, Troye-Blomberg M. Malaria blood-stage infection and its control by the immune system. Folia Biol. 1999;46(6):210–8.
- Rich SM, Leendertz FH, Xu G, Lebreton M, Djoko CF, Aminake MN, et al.The origin of malignant malaria. Proc Natl Acad Sci. 2009;106(35):14902–7.
- Wendy OM, Judith NM, Rick S, Brian G. Changes in the burden of malaria in sub-Saharan Africa. Lancet Infect Dis. 2010;10(8):545–55.
- Christopher JL, Lisa CR, Sl S, Kathryn GA, Kyle JF, Diana H, et al. Global malaria mortality between 1980 and 2010, a systematic analysis. Lancet. 2012;379:413–31.
- Louis HM, Hans CA, Xin-zhuan S, Thomas EW. Malaria biology and disease pathogenesis, insights for new treatments. Nat Med. 2013;19:156–67.
- Dauda G, Bila HA, Bashar B, Abdulrazaq S, Yahaya MS, Muhammad GM, Musa IAAMM, and Hassan HS. Exploring the Antimalarial Efficacy of Globimetula oreophila Leaf Fractions in Plasmodium berghei-infected Mice: In Vivo Approach Sciences of Phytochemistry. 2024; 3(2):105-113.
- Miller LH, Baruch DI, Marsh K, Doumbo OK. The pathogenic basis of malaria. Nature. 2002; 415:673–9.
- World Health Organization. World Malaria Report (2023); World Health Organization: Geneva, Switzerland, 2023.
- Dauda G, Jimoh Y, Akande AH, Hassan AL, Yakubu MS, Gidado I, Rabiu H, Ismail SI, Tijani OT, Olaiya AA. In-Silico Screening of Prenylated Quercetin from Globimetula oreophila Against Plasmodium falciparum Enzymes: Hope for New Antimalarial Drugs. Journal of Drug Design and Medicinal Chemistry. 2024;10(3):67-80.
- Harvey AL, Edrada-Ebal RA, Quinn RJ. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 2015, 14, 111–129.
- Schmidt TJ, Khalid SA, Romanha AJ, Alves TMA, Biavatti MW, Brun R, da Costa FB, de Castro SL, Ferreira VF, de Lacerda MVG, et al. The potential of secondary metabolites from plants as drugs or leads against protozoan neglected diseases—Part I. Curr. Med. Chem. 2012, 19, 2128–2175.
- Schmidt TJ, Khalid SA, Romanha AJ, Alves TMA, Biavatti MW, Brun R, da Costa FB, de Castro SL, Ferreira VF, de Lacerda MVG, et al. The potential of secondary metabolites from plants as drugs or leads against protozoan neglected diseases—Part II. Curr. Med. Chem. 2012, 19, 2176–2228.
- Annang F, Genilloud O, Vicente F. Contribution of natural products to drug discovery in tropical diseases. In Comprehensive Analysis of Parasite Biology: From Metabolism to Drug Discovery; Müller, S., Cerdan, R., Radulescu, O., Eds.; Wiley-VCH: Weinheim, Germany, 2016; pp. 75–104.
- Ganesan A. The impact of natural products upon modern drug discovery. Curr. Opin. Chem. Biol. 2008, 12, 306–317.
- Feher M, Schmidt JM. Property distributions: Differences between drugs, natural products, and molecules from combinatorial chemistry. J. Chem. Inform. Comput. Sci. 2003, 43, 218–227.
- Keller TH, Pichota A, Yin Z. A practical view of ‘druggability’. Curr. Opin. Chem. Biol. 2006, 10, 357–361.
- Newman DJ, Cragg GM. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335.
- Burkill HM. The useful plants of West Tropical Africa Vol. 4 Royal Botanical Gardens, Kew, Richmond UK. 1985. ISBN 9780947643010.
- Dauda G, Haruna AK, Musa AM, Hassan B, Mohammed IM, Magaji MG. In-vivo antimalarial activity of ethanol leaf extract of Globimetula oreophila (Hook. F) Danser Azadirachta Indica. BioL Environ Sci J Trop. 2016;13: 55-59.
- Dauda G, Ali BH, Muhammed SY, Muhammad MG, Ismail AM, Muhammad MA, Hassan HS. Qualitative and quantitative phytochemical profiling of ethnomedicinal folklore plant-Globimetula oreophila. J Curr Biomed Res. 2023;3:1407-1426.
- Phillips CL, Ullman B, Brennan RG. Hill, C.P. Crystal structures of adenine phosphoribosyltransferase from Leishmania donovani. EMBO J. 1999;18:3533–3545.
- Kuettel S, Greenwald J, Kostrewa D, Ahmed S, Scapozza L, Perozzo R. Crystal structures of T. b. rhodesiense adenosine kinase complexed with inhibitor and activator: Implications for catalysis and hyperactivation. PLoS Negl. Trop. Dis. 2011, 5, e1164.
- Dauda G, Ali BH, Muhammad SY, Muhammed MG, Muhammad MA, Hassan HS. Levels of trace metals content of crude ethanol leaf extract of Globimetula oreophila (Hook. f) Danser growing on Azadirachta indica using atomic absorption spectroscopy. J Curr Biomed Res. 2023;3:1397-1406.
- Dauda Garba, Bila Hassan Ali, Bashar Bawa, Abdulrazaq Sanusi, Yahaya Mohammed Sani, Muhammad Garba Magaji, Musa Isma’il Abdullahi Aliyu Muhammad Musa, and Hassan Halimatu Sadiya. Phytochemical investigation and structural elucidation of secondary metabolites from Globimetula oreophila parasitizing Azadirachta indica: A spectroscopic study (Discover plants). 2024.
- Dauda G, Haruna AK, Hassan B, Sani YM, Haruna A, Abdullahi MI, Musa AM. Prenylated Quercetin from the Leaves Extract of Globimetula Oreophila (Hook. F) Danser. Nigerian Journal of Scientific Research. 2017;16(6):725-729.
- Coombs GH, Goldberg DE, Klemba M, Berry C, Kay J, Mottram JC. Aspartic proteases of Plasmodium falciparum and other parasitic protozoa as drug targets. Trends Parasitol. 2001;17: 532-537.
- Garba D, Ali BH, Bawa B, Maryam A, Nasiru HA, Sani YM, Magaji MG, Abdullahi MI, Musa AM, Sadiya HH. Phytochemical Constituents from Globimetula oreophila as Plasmepsin I and II Inhibitors in Antimalarial Drug Discovery: An In Silico Approach. Chemistry Proceedings. 2024;16(1):42.
- Murata CE, Goldberg DE. Plasmodium falciparum falcilysin: a metalloprotease with dual specificity. J BioL Chem. 2003;278:38022-38028.
- Eggleson KK, Duffin KL, Goldberg DE. Identification and characterization of falcilysin, a metallopeptidase involved in hemoglobin catabolism within the malaria parasite Plasmodium falciparum. J Biol Chem. 1999;274:32411-32417.
- Withers-Martinez C, Jean L, Blackman MJ. Subtilisin-like proteases of the malaria parasite. Mol Microbiol. 2004;53:55-63.
- Blackman MJ. Malarial proteases and host cell egress: an 'emerging' cascade. Cell Microbiol. 2008;10:1925-1934.
- Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009;30:2785–2791.
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605-1612.
- Machin JM, Kantsadi AL, Vakonakis I. The complex of Plasmodium falciparum falcipain-2 protease with an (E)-chalcone-based inhibitor highlights a novel, small, molecule-binding site. Malar J. 2019;18:1-9.
- Kerr ID, Lee JH, Pandey KC, Harrison A, Sajid M, Rosenthal PJ, Brinen LS. Structures of falcipain-2 and falcipain-3 bound to small molecule inhibitors: implications for substrate specificity. J Med Chem. 2009;52:852-857.
- Bhaumik P, Horimoto Y, Xiao H, Miura T, Hidaka K, Kiso Y, Wlodawer A, Yada RY, Gustchina A. Crystal structures of the free and inhibited forms of plasmepsin I (PMI) from Plasmodium falciparum. J Struct BioL. 2011175:73-84.
- Asojo OA, Gulnik SV, Afonina E, Yu B, Ellman JA, Haque TS, Silva AM. Novel uncomplexed and complexed structures of plasmepsin II, an aspartic protease from Plasmodium falciparum. J Mol Biol. 2003;327:173-181.
- Smith NA, Clarke OB, Lee M, Hodder AN, Smith BJ. Structure of the Plasmodium falciparum PfSERA5 pseudo‐zymogen. Prot Sci. 2020; 29: 2245-2258.
- Lauciello L, Pernot L, Bottegoni G, Bisson W, Scapozza L, Perozzo R (To be published) P. falciparum Calcium-Dependent Protein Kinase 2 (PfCDPK2): First Crystal Structure and Virtual Ligand Screening. https://www.rcsb.org/structure/4MVF
- Kongsaeree P, Khongsuk P, Leartsakulpanich U, Chitnumsub P, Tarnchompoo B, Walkinshaw MD, Yuthavong Y. Crystal structure of dihydrofolate reductase from Plasmodium vivax: pyrimethamine displacement linked with mutation-induced resistance. Proc Natl Acad Sci. 2005;102:13046-13051.
- De Oliveira ME, Cenzi G, Nunes RR, Andrighetti CR, de Sousa Valadão DM, Dos Reis C, Simões CM, Nunes RJ, Júnior MC, Taranto AG, Sanchez BA. Antimalarial activity of 4-metoxychalcones: Docking studies as falcipain/plasmepsin inhibitors, ADMET and lipophilic efficiency analysis to identify a putative oral lead candidate. Molecules. 2013;18:15276-15287.
- Lipinski CA. Lead-and drug-like compounds: the rule-of-five revolution. Drug discovery today: Tech. 2004;1:337-341.
- Athar Alam, Manish Goyal, Mohd Shameel Iqbal, Chinmay Pal, Sumanta Dey, Samik Bindu, Pallab Maity, and Uday Bandyopadhyay. Novel antimalarial drug targets: hope for new antimalarial drugs. Expert Reviews Clinical Pharmacology. 2009;2(5):469-489.
- Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. Journal of medicinal chemistry. 2002;45(12):2615-2623.
- Li S, He H, Parthiban LJ, Yin H, Serajuddin AT. IV-IVC considerations in the development of immediate-release oral dosage form. Journal of Pharmaceutical Sciences. 2005;94(7):1396-1417.
- Maximo MdS, Comin M, Thiago SD, Mary AF, Joao EC, Maria CV, Anelise SNF. Synthesis, antiproliferative activity and molecular properties predictions of galloyl derivatives. Molecules. 2015;20(4):5360-5373.
- Samuel BB, Oluyemi WM, Johnson TO, Adegboyega AE. High-Throughput Virtual Screening with Molecular Docking, Pharmacophore Modelling and ADME Prediction to Discover Potential Inhibitors of Plasmodium falciparum Lactate Dehydrogenase (PfLDH) from Compounds of Combretaceae Family. Tropical Journal of Natural Product Research. 2021;5(9):1665-1672.
- Ojo OA, Ojo AB, Okolie C, Abdurrahman J, Arnabas M, Evbuomwan IO, et al. Elucidating the interactions of compounds identified from Aframomum melegueta seeds as promising candidates for the management of diabetes mellitus: A computational approach. Informatics in Medicine Unlocked. 2021;26:100720.
- Huang SM, Strong JM, Zhang L, Reynolds KS, Nallani S, Temple R, Chung S. New era in drug interaction evaluation: US Food and Drug Administration update on CYP enzymes, transporters, and the guidance process. J Clin Pharm. 2008; 48(6):662-670.
- Li X, Chen L, Cheng F, Wu Z, Bian H, Xu C, Li W, Liu G, Shen X, Tang Y. In Silico Prediction of Chemical Acute Oral Toxicity Using Multi-Classification Methods. J Chem Informat Model. 2014;54(4):1061-1069.
- Banerjee P, Kemmler E, Dunkel M, Preissner R. ProTox 3.0: a web server for the prediction of toxicity of chemicals. Nucleic Acids Research. 2024;gkae303.
- Akar S, Çırak T, Hüsunet MT, Turkdonmez I, Kenger İH, Aslan F, Kardöl A, Yıldız H, Zencir S, Emek AG, Kayraldız A. Determination of mutagenic potentials of diarylmethylamine based imine compounds by ames test and computational molecular docking. Exp AppL Med Sci. 2003;4:610-620.