
RESEARCH ARTICLE
In-silico study for African plants with possible beta-cell regeneration effect through inhibition of DYRK1A
Academic Editor: James H. Zothantluanga
Sciences of Phytochemistry|Vol. 1, Issue 1, pp. 11-25 (2022)
Views
Downloads
Shares
Received
Jun 25, 2022Revised
Jul 1, 2022Accepted
Jul 1, 2022Published
Jul 1, 2022
Abstract
The continuous destruction of normal insulin-producing pancreatic beta-cells is a contributing factor in all common forms of diabetes, due to insufficient production of insulin, especially in type 1 diabetes. There are attempts at beta-cells transplantation, but the cost and availability of donors pose a great challenge to the process. Dual-Specificity Tyrosine Phosphorylation-Regulated Kinase A (DYRK1A) plays a crucial role in beta-cells destruction. Our research targets to identify plants that can be utilized as a possible alternative approach to beta-cell replacement through a pharmacologically induced regeneration of new beta-cells in-silico. The 3D structure DYRK1A and 6511 phytochemicals were obtained from the Protein Data Bank and the African Natural Products Database respectively. They were duly prepared for molecular docking simulations (MDS). MDS was implemented, after validation of docking protocols, in AutoDock-Vina®, with virtual screening scripts. Phytocompounds with good binding affinities for DYRK1A were selected as frontrunners. The compounds were screened for toxicity, Lipinski’s rule confirmation with Data Warrior software followed by kinase inhibitory bioactivity prediction with the Molinspiration Chemoinformatics web tool. Twelve phytocompounds were found to be predictably highly active in-silico against DYRK1A with good drug-like property based on Lipinski’s rule, non-mutagenic, non-tumorigenic, no reproductive effect, and non-irritant, with high predicted bioactivity. In-silico active phytocompounds against DYRK1A with their plant sources and physicochemical parameters were identified. Further studies will be carried out in-vitro and in-vivo to validate the results of this study using plants containing the identified phytocompounds.
Introduction
Diabetes is a life-threatening global health issue as a result of its high incidence (1), associated disability, and mortality (2). The pancreatic beta-cell deficit is a significant part of the pathophysiological mechanism (3). Beta-cells considerable damage leads to long-lasting endocrine insufficiency with the possibility of a permanent diabetic state. On the bright side, pancreatic beta-cell regeneration is a promising pharmacological strategy for recovering Beta-cells. In adults, it is known that the endocrine pancreas has a regulated ability for self-regeneration (4). Consequently, approaches for stimulating beta-cell restoration have insightful inferences for the treatment and management of diabetes, particularly for type 1 diabetes and late-type 2 diabetes with considerable beta-cell loss.
Two possible approaches exist through which pancreatic beta-cells can be regenerated. The first approach is by preventing beta-cell loss precisely through the inhibition of beta-cell apoptosis and dedifferentiation. The second approach is to stimulate new endogenous regeneration and exogenous supplementation. For about a century, researchers have attempted pancreatic beta-cells regeneration. Under specific physiological environments, such as pregnancy, obesity, and conditions of insulin resistance, the adaption of islet and improved beta-cell mass take place in animal models and humans (5-8). Contemporary advances in new technologies have offered additional substantiation on the generation of beta-cells. Single-cell RNA sequencing available data have revealed that human islets comprise four discrete subtypes of beta-cells (9) and probably transitional phases (10). These suggest that beta-cells can acclimatize and undergo transdifferentiation or neogenesis. Physiological restoration research can make available data on the development of medication targeted toward beta-cell regeneration. Several approaches have been reported to be utilized in the promotion of beta-cells regeneration. The strategies include pancreatectomy, partial duct ligation, and chemical-induced massive beta-cell loss (11-15). Molecular routes that cause multiplications in the mass of beta-cells have been comprehensively explored. Only a small portion of the materials studied have been found to have clinical, pre-clinical, or clinical potential as medicines. Thousands of materials have been studied, and hundreds are effective in the course of beta-cell restoration.
The CMGC (CDK, MAPK, CDC-like kinases, GSK3 kinase) family of eukaryotic protein kinases has been demonstrated to play crucial roles in neurodegenerative illnesses (16, 17), cell death, and tumorigenesis. Dual-specificity tyrosine phosphorylation-regulated kinase A (DYRK1A) is a member of this family (18, 19). A regulator of regeneration pathways essential to human insulin-producing pancreatic -cells has recently been discovered as DYRK1A (20-23). Numerous studies have explored the development of DYRK1A inhibitor scaffolds, given the involvement of DYRK1A in these diseases (17-20, 22-24). Several DYRK1A inhibitors from natural sources like harmine and small molecules have been identified and characterized (22, 25-48). Harmine and its analogs (-carbolines) are the most often researched DYRK1A inhibitors, and they continue to be among the most effective and readily available inhibitor families that can be taken orally (17, 49). The presence of harmine in the hallucinogenic infusion of ayahuasca and its affinity for serotonin, tryptamine, and other receptors in the central nervous system, in addition to its kinase inhibitory activity, have led to the hypothesis that harmine is a hallucinogen (50, 51). Harmine and its analogs had been reported to block the DYRK1A-mediated phosphorylation of tau proteins in the CNS (52). They also showed anti-proliferative action, including inhibition of topoisomerase I (53, 54), inhibition of CDKs (55), activation of cell apoptosis (56), and DNA intercalation (57).
This research aims to determine druggable enzyme/target/receptor that is vital in the pathogenesis of beta-cell apoptosis, identify phytocompounds with high binding affinity against the identified target using molecular docking simulation, and determine the drug-likeness of promising phytocompounds based on Lipinski’s rule, determine the toxicity of the phytocompounds in-silico, undertake bioactivity prediction of the phytocompounds on Molinspiration platform and identify the plant sources of the frontrunner compounds.
Experimental Section
Materials
Personal computer, African Natural Compounds Database, PubChem (http://Pubchem. ncbi. nlm. nih. gov) (58), Linux operating system (Ubuntu desktop 18.04), Protein data bank (https://www. rcsb. org/) (59), DataWarrior software (60), PyMol software (61), AutoDockTools-1.5.6 software (62), Autodockvina 1.1.2 software (63), on Ubuntu operating system, Molinspiration Chemoinformatics web tool (https://www.molinspiration.com/cgi-bin/properties) (64).
Literature Mining
Literature was mined to identify the target/receptor for possible induction of beta-cell regeneration. This was done to check the importance of the target/receptors in the onset and pathophysiology of beta-cell destruction. This gives more information about the receptor, functions, properties, and its druggability.
Selection and Preparation of the Receptor
After the identification of several targets/receptors, literature mining, and analysis of the target/receptor, Dual-specificity tyrosine phosphorylation-regulated kinase A in 3D format was obtained from Protein Data Bank (PDB) with the respective PDB code; 6UWY. The initial preparation of the PDB file to select the required chains, and delete multiple ligands was done with PyMol software. The PyMol software was employed to gain insight into the ligands binding to the receptors. The receptor was prepared for molecular docking simulations with the AutoDockTool. In the preparation, polar hydrogens and Kollman’s charges were added to the receptors and they were saved in the pdbqt file format. The pdbqt file format is the structural format required for the protein and ligand for carrying out molecular docking simulation. The electrostatic grid boxes and the 3-dimensional affinity of different sizes and centers, as indicated in Table 1 below, were created around the active site of the protein.
| Axis | Centres | Sizes |
|---|---|---|
| X | -59.224 | 10 |
| Y | -24.052 | 8 |
| Z | 24.659 | 12 |
Selection, Drug-likeness, and Toxicity Assessment of Ligands (Phytocompounds)
A total number of 6511 isolated phytocompounds were obtained from the African Natural Products Database (African-compounds. org) (65, 66) in the 3D-structure data file format. The phytocompounds were loaded onto the DataWarrior software. Molecular properties such as molecular weight, hydrogen bond donor, hydrogen bond acceptor, partition coefficient (log P), and topological polar surface area (TPSA) were determined. Lipinski's rule of five violations was noted. The phytocompounds were also screened for toxicity (mutagenicity, carcinogenicity, tumorigenicity, and reproductive effect).
Selection and Preparation of Ligands
Phytocompounds following Lipinski’s rule of 5 with no toxicity in-silico were prepared for the molecular docking simulation. Reference ligands were identified from the literature including the compound co-crystallized with the receptor/protein on the PDB database. In preparation of the ligands for molecular docking simulation, all rotatable bonds, Torsions, and Geistegers charges were assigned and saved in the pdbqt file format.
Validation of Docking Protocol
To validate the molecular docking simulations protocol for the 6UWY (DYRK1A) protein, the PDB structure of this protein in complex with a reference inhibitor was reproduced in-silico. The deletion of the reference compound from the protein was done with the PyMol software. Polar hydrogen, Kollman charges, grid box sizes, and centers at a grid space of 1.0 Å were determined with the AutoDockTools-1.5.6 (62, 63). The protein was saved in the pdbqt file format. The reference compound was prepared for molecular docking simulation with the AutoDockTools-1.5.6. All rotatable bonds, including torsions, were permitted to remain rotatable. Then, output was produced with the pdbqt file extension. Molecular docking simulation of the protein and reference compound was implemented locally with the AutoDockVina® (63) on a Linux platform using the centers and sizes with a virtual screening shell script. Docked conformations were visualized in the PyMol-1.4.1 software and the binding poses of the co-crystal inhibitors were compared with the re-docked co-crystal structures of the reference compound.
Molecular Docking of the Phytocompounds on Dual-Specificity Tyrosine Phosphorylation-Regulated Kinase A
The phytocompounds were prepared in batches for molecular docking simulations using virtual screening scripts against the dual-specificity tyrosine phosphorylation-regulated kinase A. Following the validation of docking methods, four replicates of Molecular Docking Simulations were performed on a Linux platform using AutoDockVina® and related tools. To determine the leading phytocompounds, binding free energy values (kcal/mol SD) were ranked.
Bioactivity Prediction of Phytocompounds
The online Molinspiration web tool version 2011.06 (www.molinspiration.com) was supplied SMILES notations of the leading phytocompounds to forecast the bioactivity scores for kinase inhibition.
Calculation of the Predicted Percentage of Absorption
The predicted percentage of absorption (%ab) of the frontrunner phytocompounds was calculated with the method reported by Zhao et al. (2002) (67). The following formula was used: %ab = 109 – (0.345 x TPSA).
Results
Drug-Likeness, and Toxicity Assessment of Ligands (Phytocompounds)
The drug-likeness assessment of the 6511 phytocompounds based on Lipinski’s rule of five was done to screen out phytocompounds with violations of the rules. After the screening, a total number of 3814 phytocompounds had no violation of Lipinski’s rule, while 2697 phytocompounds violated the rules. Toxicity assessment on the 3814 phytocompounds that did not violate Lipinski's rule was carried out with DataWarrior in to identify phytocompounds that might be mutagenic, tumorigenic, irritating, or have reproductive consequences. A total number of 1897 phytocompounds were found to have none of the listed toxicities in-silico. Total polar surface area (TPSA) was also analyzed for all the phytocompounds.
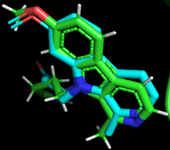
Validation of Docking Protocol
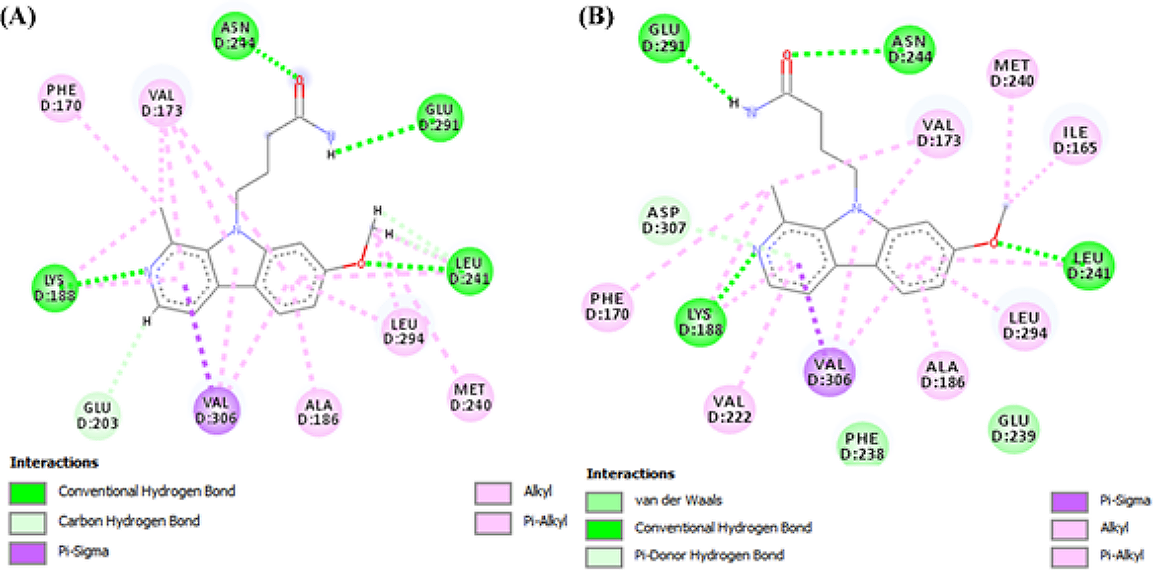
The docking protocol validation was done to ensure in-silico reproducibility of the experimental protein-ligand interactions obtained from the protein data bank. The results obtained from the docking validations are presented below in Figures 1 and 2. Figure 1 represents the structural conformation and superimposition of the docked ligand (blue) and co-crystallized ligand (green) in the Dual-specificity tyrosine phosphorylation-regulated kinase A binding site. Figure 2A shows the 2D representation of the co-crystallized ligand-protein interaction, while Figure 2B shows the 2D representation of the docked ligand-protein interaction. Comparative analysis of the docked ligand and co-crystallized ligand-protein interaction reveals a 90.9% match.
Molecular Docking of the Phytocompounds DYKR1A Protein
The molecular docking of the phytocompounds was performed on DYKR1A to identify phytocompounds with better in-silico inhibitory activity against DYKR1A than the reference compounds. The reference compounds are listed in the last three rows of Table 2. The docking was also performed to study the phytocompounds-proteins interaction pattern at the binding sites of these proteins. Phytocompounds with better binding affinities/energies than the reference compounds as can be observed from the mean binding affinity, are presented in Table 2.
| S/N | Compound Name | Mean binding affinity | Molecular Weight | cLogP | Hydrogen Acceptor | Hydrogen Donor | TPSA |
|---|---|---|---|---|---|---|---|
| 1 | Lanuginosine | -11.3 ± 0 | 305.29 | 3.46 | 5 | 0 | 57.65 |
| 2 | 4-Beta, 8-alpha-dihydroxy-6-alpha-vanilloyloxydauc-9-ene | -11.23 ± 0.06 | 400.51 | 3.25 | 5 | 1 | 72.83 |
| 3 | Aegyptinone A | -10.87 ± 0.06 | 310.39 | 1.29 | 3 | 0 | 57.20 |
| 4 | Sigmoidin A | -10.70 ± 0.17 | 424.49 | 5.86 | 6 | 4 | 107.22 |
| 5 | Penilactone | -10.70 ± 0.00 | 304.3 | 1.67 | 6 | 1 | 89.90 |
| 6 | Altertoxin I | -10.60 ± 0.00 | 352.34 | 2.36 | 6 | 4 | 115.06 |
| 7 | Sigmoidin B | -10.50 ± 0.00 | 356.37 | 3.83 | 6 | 4 | 107.22 |
| 8 | 6, 7-Dehydro-19-beta-hydroxyschizozygin | -10.50 ± 0.00 | 337.4 | 0.53 | 5 | 1 | 43.21 |
| 9 | Ungeremine | -10.40 ± 0.00 | 265.27 | 3.42 | 4 | 1 | 43.62 |
| 10 | Anastatin B | -10.40 ± 0.00 | 378.34 | 3.58 | 7 | 4 | 120.36 |
| 11 | Latrunculin B | -10.40 ± 0.00 | 357.56 | 4.49 | 4 | 2 | 83.86 |
| 12 | Scalarolide | -10.40 ± 0.00 | 386.57 | 4.51 | 3 | 1 | 46.53 |
| 13 | Feselol | -10.40 ± 0.00 | 386.53 | 3.61 | 4 | 1 | 55.76 |
| 14 | Assafoetidnol A | -10.40 ± 0.00 | 398.5 | 3.15 | 5 | 2 | 75.99 |
| 15 | Chamanetin | -10.40 ± 0.00 | 364.4 | 3.80 | 5 | 3 | 86.99 |
| 16 | Neoclerodan-5, 10-en-19, 6beta, 20, 12-diolide | -10.40 ± 0.00 | 315.48 | 1.96 | 2 | 0 | 40.13 |
| 17 | Chrysophanol- isophyscion bianthrone | -10.37 ± 0.06 | 508.53 | 4.63 | 7 | 4 | 124.29 |
| 18 | 3-Taraxasterol | -10.30 ± 0.00 | 430.76 | 9.48 | 1 | 1 | 20.23 |
| 19 | Helioscopinolide C | -10.30 ± 0.00 | 330.42 | 2.43 | 4 | 1 | 63.60 |
| 20 | 3beta-hydroxyisopimaric acid | -10.30 ± 0.00 | 317.45 | 1.40 | 3 | 1 | 60.36 |
| 21 | Taraxasterol | -10.23 ± 0.06 | 424.71 | 7.00 | 1 | 1 | 20.23 |
| 22 | 3beta-hydroxymansumbin-13 (17)-en-16-one | -10.20 ± 0.00 | 332.53 | 4.53 | 2 | 1 | 37.30 |
| 23 | Dihydrofumariline | -10.20 ± 0.00 | 354.38 | 1.15 | 6 | 2 | 61.59 |
| 24 | 12alpha-acetoxy-24, 25-epoxy-24-hydroxy-20, 24-dimethylscalarane | -10.17 ± 0.35 | 460.7 | 5.86 | 4 | 1 | 55.76 |
| 25 | 3, 4, 18-cyclopropa-12-hydroxy-ent-abiet-7-en-16, 14-olide | -10.13 ± 0.06 | 316.44 | 2.70 | 3 | 1 | 46.53 |
| 26 | 13-Hydroxyfeselol | -10.13 ± 0.06 | 400.51 | 3.53 | 5 | 2 | 75.99 |
| 27 | Stemmin C | -10.10 ± 0.00 | 332.48 | 3.40 | 3 | 2 | 57.53 |
| 28 | Helioscopinolide A | -10.10 ± 0.00 | 318.46 | 3.07 | 3 | 1 | 46.53 |
| 29 | Foetidin | -10.10 ± 0.17 | 381.49 | 5.47 | 4 | 2 | 51.83 |
| 30 | 2, 11-didehydro-2- dehydroxylycorine | -10.10 ± 0.00 | 274.34 | 0.05 | 4 | 2 | 43.13 |
| 31 | Voucapane | -10.10 ± 0.00 | 286.46 | 5.48 | 1 | 0 | 13.14 |
| 32 | Trachyloban-19-oic Acid | -10.10 ± 0.00 | 299.43 | 1.42 | 2 | 0 | 40.13 |
| 33 | Abyssinin II | -10.10 ± 0.10 | 370.4 | 4.11 | 6 | 3 | 96.22 |
| 34 | (-)-Semiglabrin | -10.10 ± 0.00 | 392.41 | 4.24 | 6 | 0 | 71.06 |
| 35 | Taraxerone | -10.10 ± 0.61 | 426.73 | 7.59 | 1 | 0 | 17.07 |
| 36 | Pratorinine | -10.07 ± 0.06 | 267.28 | 2.63 | 4 | 1 | 49.77 |
| 37 | Ergosterol | -10.07 ± 0.92 | 396.66 | 6.87 | 1 | 1 | 20.23 |
| 38 | Solanidin | -10.07 ± 0.06 | 400.67 | 3.2 | 2 | 2 | 24.67 |
| 39 | Calotroproceryl acetate B | -10.00 ± 0.00 | 466.75 | 7.66 | 2 | 0 | 26.30 |
| 40 | Botryorhodine B | -10.00 ± 0.00 | 314.29 | 3.45 | 6 | 2 | 93.06 |
| 41 | Asteriscunolide A | -10.00 ± 0.00 | 250.34 | 2.93 | 3 | 0 | 43.37 |
| 42 | Diazo derivative of Inuloxin A | -10.00 ± 0.00 | 264.36 | 3.61 | 3 | 0 | 35.53 |
| 43 | Thymelol | -10.00 ± 0.00 | 354.31 | 1.87 | 7 | 1 | 91.29 |
| 44 | Polyanthin | -10.00 ± 0.69 | 424.54 | 4.92 | 5 | 0 | 61.83 |
| 45 | Samarcandin | -10.00 ± 0.44 | 400.51 | 3.76 | 5 | 2 | 75.99 |
| 46 | 8alpha-isobutanoyloxy-5-Alpha-Hydroxy-2- Oxo-11, 13-dehydroguaia-1 (10), 3-dien-6alpha, 12-Olide | -10.00 ± 0.00 | 334.41 | 1.85 | 5 | 1 | 72.83 |
| 47 | Aloenin acetal | -10.00 ± 0.00 | 436.41 | 0.33 | 10 | 3 | 133.14 |
| 48 | Retroisosenine | -10.00 ± 0.00 | 336.41 | -0.99 | 6 | 1 | 66.27 |
| 49 | Ent-trachyloban-18- oic Acid | -10.00 ± 0.00 | 301.45 | 1.69 | 2 | 0 | 40.13 |
| 50 | Trachylobane | -10.00 ± 0.00 | 274.49 | 5.48 | 0 | 0 | 0.00 |
| 51 | Lanceolatin B | -10.00 ± 0.00 | 262.26 | 3.82 | 3 | 0 | 39.44 |
| 52 | 12-Hydroxy-8, 12-Abietadiene-3, 11, 14-Trione | -10.00 ± 0.00 | 329.42 | 1.05 | 4 | 0 | 74.27 |
| 53 | Hosloppone | -10.00 ± 0.00 | 300.44 | 4.41 | 2 | 2 | 40.46 |
| 54 | Abyssinone II | -10.00 ± 0.00 | 324.38 | 4.52 | 4 | 2 | 66.76 |
| 55 | Lanceolatin A | -9.97 ± 0.40 | 336.39 | 4.21 | 4 | 1 | 55.76 |
| 56 | Postratol | -9.97 ± 0.06 | 460.61 | 8.57 | 4 | 2 | 66.76 |
| 57 | Erythroxyl-4 (17), 15 (16)-Dien-3-One | -9.97 ± 0.06 | 270.41 | 4.54 | 1 | 0 | 17.07 |
| 58 | 3-O-Benzoylhosloppone | -9.97 ± 0.12 | 420.55 | 4.76 | 4 | 1 | 63.60 |
| 59 | 7-Keto-8alpha-hydroxy-deepoxysarcophine | -9.93 ± 0.06 | 332.44 | 3.49 | 4 | 1 | 63.60 |
| 60 | 3-[6-(3-Methyl-But-2-enyl)-1H-Indolyl]-6-(3-methyl-but-2-enyl)-1H-Indole | -9.93 ± 0.06 | 368.52 | 7.25 | 2 | 1 | 20.72 |
| 61 | (6Z)-Cladiellin (cladiella-6Z, 11 (17)-dien-3-Ol) | -9.90 ± 0.00 | 306.49 | 4.64 | 2 | 1 | 29.46 |
| 62 | Hippacine | -9.90 ± 0.00 | 251.24 | 2.78 | 4 | 2 | 62.46 |
| 63 | 1, 2-Dehydrobeninine | -9.90 ± 0.00 | 327.45 | -0.34 | 4 | 2 | 34.93 |
| 64 | Sipholenol J | -9.90 ± 0.00 | 462.67 | 4.13 | 5 | 3 | 86.99 |
| 65 | Wtmannin | -9.90 ± 0.00 | 428.44 | 1.62 | 8 | 0 | 109.11 |
| 66 | Gummosin | -9.90 ± 0.00 | 384.51 | 3.58 | 4 | 1 | 55.76 |
| 67 | Badrakemin | -9.90 ± 0.35 | 382.54 | 4.98 | 3 | 2 | 38.69 |
| 68 | (-)-Samarcandone | -9.90 ± 0.00 | 398.5 | 3.78 | 5 | 2 | 72.83 |
| 69 | Totaradiol | -9.90 ± 0.00 | 302.46 | 4.52 | 2 | 2 | 40.46 |
| 70 | Abietatriene | -9.90 ± 0.00 | 268.44 | 5.55 | 0 | 0 | 0.00 |
| 71 | 6, 7-Dehydroroyleanon | -9.90 ± 0.00 | 313.42 | 1.48 | 3 | 0 | 57.2 |
| 72 | 5-OH-3-methylnaphtho [2-3-C]Furan-4, 9-dione | -9.90 ± 0.00 | 232.23 | 1.40 | 4 | 1 | 67.51 |
| 73 | 3'-Prenylnaringenin | -9.90 ± 0.00 | 338.36 | 4.36 | 5 | 3 | 86.99 |
| 74 | Lysicamine | -9.9 ± 0. | 291.31 | 3.28 | 4 | 0 | 48.42 |
| 75 | 5-Deoxyabyssinin II | -9.87 ± 0.15 | 354.4 | 4.45 | 5 | 2 | 75.99 |
| 76 | Ekeberin A | -9.87 ± 0.06 | 456.71 | 6.19 | 3 | 0 | 35.53 |
| 77 | Aegyptinone B | -9.83 ± 0.06 | 327.4 | 1 | 4 | 1 | 77.43 |
| 78 | Pratorimine | -9.8 ± 0 | 265.27 | 3.06 | 4 | 1 | 51.46 |
| 79 | Anhydroverlotorin | -9.8 ± 0 | 250.34 | 3.09 | 3 | 0 | 43.37 |
| 80 | Nagilactone F | -9.8 ± 0 | 316.4 | 2.2 | 4 | 0 | 52.60 |
| 81 | Totarolone | -9.8 ± 0 | 300.44 | 4.66 | 2 | 1 | 37.30 |
| 82 | Voucapan-5-ol | -9.8 ± 0 | 300.44 | 4.38 | 2 | 1 | 33.37 |
| 83 | Coladonin | -9.8 ± 0.82 | 384.51 | 3.93 | 4 | 1 | 55.76 |
| 84 | Anhydrolycorine | -9.8 ± 0.17 | 251.28 | 2.98 | 3 | 0 | 21.70 |
| 85 | 8-C-P-Hydroxybenzylluteolin | -9.8 ± 0.69 | 392.36 | 3.56 | 7 | 5 | 124.29 |
| 86 | 4-(7-Methoxy-1-methyl-9H-beta-carbolin-9-Yl)butanamide | -9.80 ± 0.00 | 297.36 | 1.97 | 5 | 2 | 70.15 |
| 87 | (1Z)-1-(3-Ethyl-5-hydroxy-2 (3H)-benzothiazolylidene)-2-propanone (INDY) | -7.50 ± 0.00 | 235.31 | 2.01 | 3 | 1 | 42.23 |
| 88 | Gnf4877 | -7.28 ± 0.10 | 494.53 | 2.51 | 10 | 4 | 143.57 |
Bioactivity Prediction of Phytocompounds
Results of the bioactivity prediction of the 85 phytocompounds with better binding affinities than the reference compounds are presented in Table 3. The phytocompounds were screened for kinase inhibitory activity because the protein of interest (DYRK1A) is a kinase. Twelve phytocompounds were found to possess kinase inhibitory activity based on the scores. Some of the phytocompounds have better inhibitory scores than the reference compounds as can be observed in Table 3.
| S/N | Phytocompounds | Kinase inhibitoryscore | Plant sources |
|---|---|---|---|
| 1 | Lysicamine | 0.42 | Annickia kummeriae |
| 2 | Lanuginosine | 0.40 | Magnolia grandiflora |
| 3 | Pratorinine | 0.40 | Crinum americanum |
| 4 | Hippacine | 0.40 | Crinum bulbispermum |
| 5 | Pratorimine | 0.40 | Crinum americanum |
| 6 | 4-(7-methoxy-1-methyl-9-H-beta-carbolin-9-yl)-butanamide | 0.37 | |
| 7 | 3-[6-(3-methyl-but-2-enyl)-1-H-indolyl]-6-(3-methyl-but-2-enyl)-1-H-indole | 0.32 | Monodora angolensis |
| 8 | 8-C-p-hydroxybenzylluteolin | 0.27 | Thymus hirtus |
| 9 | GNF4877 | 0.25 | |
| 10 | 3'-Prenylnaringenin | 0.21 | Erythrina abyssinica |
| 11 | Lanceolatin B | 0.15 | Tephrosia purpurea |
| 12 | Lanceolatin A | 0.10 | Tephrosia purpurea |
| 13 | Aegyptinone B | 0.02 | Zhumeria majdae |
| 14 | (-)-Semiglabrin | 0.00 | Tephrosia purpurea |
| 15 | (1Z)-1-(3-Ethyl-5-hydroxy-2-(3H)-benzothiazolylidene)-2-propanone (INDY) | -0.47 |
Calculation of the Predicted Percentage of Absorption
The results of the predicted percentage absorption of the frontrunner phytocompounds with that of the reference compounds are presented in Table 4. The prediction is based on the TPSA values.
Discussion
The study was set out to determine the binding affinities of phytocompounds from the African natural product database to DYRK1A compared to the reference compounds INDY, 4-(7-methoxy-1-methyl-9H-beta-carbolin-9-yl) butanamide and GNF4877 with in-silico molecular docking simulation. The process of developing new drugs has proven to be difficult due to the enormous expense of drug discovery and development, and the time needed. Drug discovery and development are sped up by computer-aided drug design elements including molecular docking, molecular dynamics, QSAR, ADMET tool, and their accurate predictions.
| Compounds | TPSA | %Ab |
|---|---|---|
| 3-[6-(3-methyl-but-2-enyl)-1-H-indolyl]-6-(3-methyl-but-2-enyl)-1-H-indole | 20.72 | 101.85 |
| Lanceolatin B | 39.44 | 95.39 |
| (1Z)-1-(3-Ethyl-5-hydroxy-2 (3H)-benzothiazolylidene)-2-propanone (INDY) | 42.23 | 94.43 |
| Lysicamine | 48.42 | 92.30 |
| Pratorinine | 49.77 | 91.83 |
| Pratorimine | 51.46 | 91.25 |
| Lanceolatin A | 55.76 | 89.76 |
| Lanuginosine | 57.65 | 89.11 |
| Hippacine | 62.46 | 87.45 |
| 4-(7-methoxy-1-methyl-9-H-beta-carbolin-9-yl)-butanamide | 70.15 | 84.80 |
| (-)-Semiglabrin | 71.06 | 84.48 |
| Aegyptinone B | 77.43 | 82.29 |
| 3'-Prenylnaringenin | 86.99 | 78.99 |
| 8-C-p-hydroxybenzylluteolin | 124.29 | 66.12 |
| GNF4877 | 143.57 | 59.47 |
On the other hand, medicines and medicinal substances have historically been derived from nature, primarily plants. The majority of medicines on the market today are either isolated or created from isolates derived from natural sources. Based on their use in conventional medical procedures, the majority of these currently utilized medications are made from natural sources (68). To date, more novel compounds are being isolated from plants (69) and deposited in chemical databases. There are also general biological databases and specialized databases on which thousands of proteins are deposited to aid scientific research (70).
In this work, we retrieved 6511 phytocompounds from the African natural database that were purported to be isolated from African plants. Lipinski's rule of five was utilized to determine the drug-likeness of these phytocompounds. Pharmaceutical chemists frequently utilize Lipinski's rule of five to forecast the oral bioavailability of possible lead or therapeutic compounds during drug design and development. Lipinski's "rule of five" states that a candidate molecule is more likely to be orally active if it meets the following criteria: a) has a molecular weight below 500; b) has an estimated octanol/water partition coefficient (Log P is less than 5); c) has fewer than five hydrogen bond donors; and d) has fewer than ten hydrogen bond acceptors (71-73).
The DataWarrior software uses a precomputed collection of structural pieces that, when found in the structures under study, trigger toxicity alerts. All compounds from the Registry of Toxic Effects of Chemical Substances (RTECS) database (74) that are acknowledged to be active in a certain toxicity class were thoroughly destroyed to compile these fragment lists. During the shredding, compounds were first severed, with each rotating link leading to a set of core fragments. These, in turn, were utilized to reconstitute all possible significant substructures of the original molecule. The frequency of any fragment (core and created fragments) within all chemicals in that toxicity class was then determined using a substructure search process. Additionally, it identified these fragment frequencies in more than 3000 traded medications' structural data. Any fragment was viewed as a risk factor if it frequently occurs as the substructure of dangerous chemicals but never or very occasionally in the traded pharmaceuticals. This assumption was made based on the notion that sold drugs are primarily free of toxic effects. A total of 1897 phytocompounds did not exhibit any in-silico mutagenicity, tumorigenic, irritant, or reproductive impacts based on this explained fragments search. No fragments or fragments known to have any of the toxicities listed in the Registry of Toxic Effects of Chemical Substances were present in these phytocompounds.
From the molecular docking result, 85 phytocompounds were obtained with better binding affinity than the reference compounds, as shown in Table 2. Lower binding affinity suggests better ligand binding. The importance of binding affinity values is determined by the most significant magnitude negative value, representing the most favorable conformation of the complex formed when the ligand involved efficiently binds with the protein's active site. As observed, the mean binding affinity scores are in negative values. This is because protein-ligand binding only occurs spontaneously when the free energy change is negative, and the difference in ∆G levels of complexed and unbound free states is proportional to the stability of the protein-ligand interaction. Both protein folding and protein-ligand binding occur when ∆G is low in the system (75, 76). Hence, negative ∆G scores indicate the stability of the resulting complexes with the receptor molecules, which is an essential characteristic of efficacious drugs (77).
From the molinspiration bioactivity prediction, twelve compounds were found to be very active kinase inhibitors. Based on the prediction, two of the three reference compounds used were also very active kinase inhibitors. One of the reference compounds was predicted to be a moderately active kinase inhibitor. In molinspiration, biological activity is measured by a bioactivity score that is categorized as active (0.00 to 0.5), moderately active (0.00 to -0.5), and inactive (less than -0.5) (64).
The calculated percentage absorption (%ABS) of the frontrunner phytocompounds ranged between 66.12% and 101.85%, indicating that these phytocompounds have good permeability in the cellular membrane. The percentage absorption was calculated from the topological polar surface area (TPSA). The frontrunner phytocompounds exhibited computational TPSA values between 20.72 and 124.29 Å2 and have good intestinal absorption. As a guide, orally active drugs transported by the transcellular route should not exceed a PSA of about 120 Å2 (78, 79). Similarly, for good brain penetration of CNS drugs, this number should even be tailored to PSA< 100 Å2 (79) or even smaller, < 60–70 Å2 (78).
Finally, observation of the frontrunner phytocompounds' structures compared with reference compounds, as presented in Figure 3, reveals some structural activity relationships that might be necessary for the inhibition of DYRK1A. The frontrunner compounds are composed of phenolics and alkaloids. From the 2D structure of the PDB reference compound presented in Figure 2, it can be observed that nitrogen, oxygen, and hydrogen atoms (which are all components of the frontrunner phytocompounds) are necessary for the protein-ligand interactions. Previous in-vitro research has shown that some natural products such as alkaloids and polyphenolic compounds act as inhibitors of DYRK1A. Epigallocatechin gallate, a major catechin component of green tea, when tested in a panel of 28 kinases structurally and functionally related to DYRK1A, showed selective inhibitory activity against DYRK1A (IC50 330 nM [ATP] = 100 μM) (80). Acaninol B, which was isolated from the Leguminosae plant Acacia nilotica (81), exhibited only moderate action against DYRK1A (IC50 19 M [ATP] = 15 M) (82). The already known CDK inhibitor flavopiridol was discovered to be a powerful DYRK1A inhibitor (IC50 0.3 M) (85) after being screened against a panel of five kinases using a set of natural and synthetic flavonoids and flavonoidal alkaloids. A strong DYRK1A inhibitor with an IC50 of 19 nM, staurosporine is an indolecarbazole derived from Streptomyces staurosporeus (86). However, it is highly nonselective toward other kinases (87, 88). The L-rhamnulose-modified staurosporine analog was similarly significantly effective against DYRK1A (IC50 4 nM) (88). Alkaloid acrifoline has a very strong DYRK1A inhibitory effect (IC50 = 0.075 M). Atalaphyllidine and chlorospermine B are both moderately effective DYRK1A inhibitors (89). Recent studies have demonstrated the potency of two granulatimide analogs as DYRK1A inhibitors, with IC50 values of 0.26 and 0.09 M, respectively (90, 91).
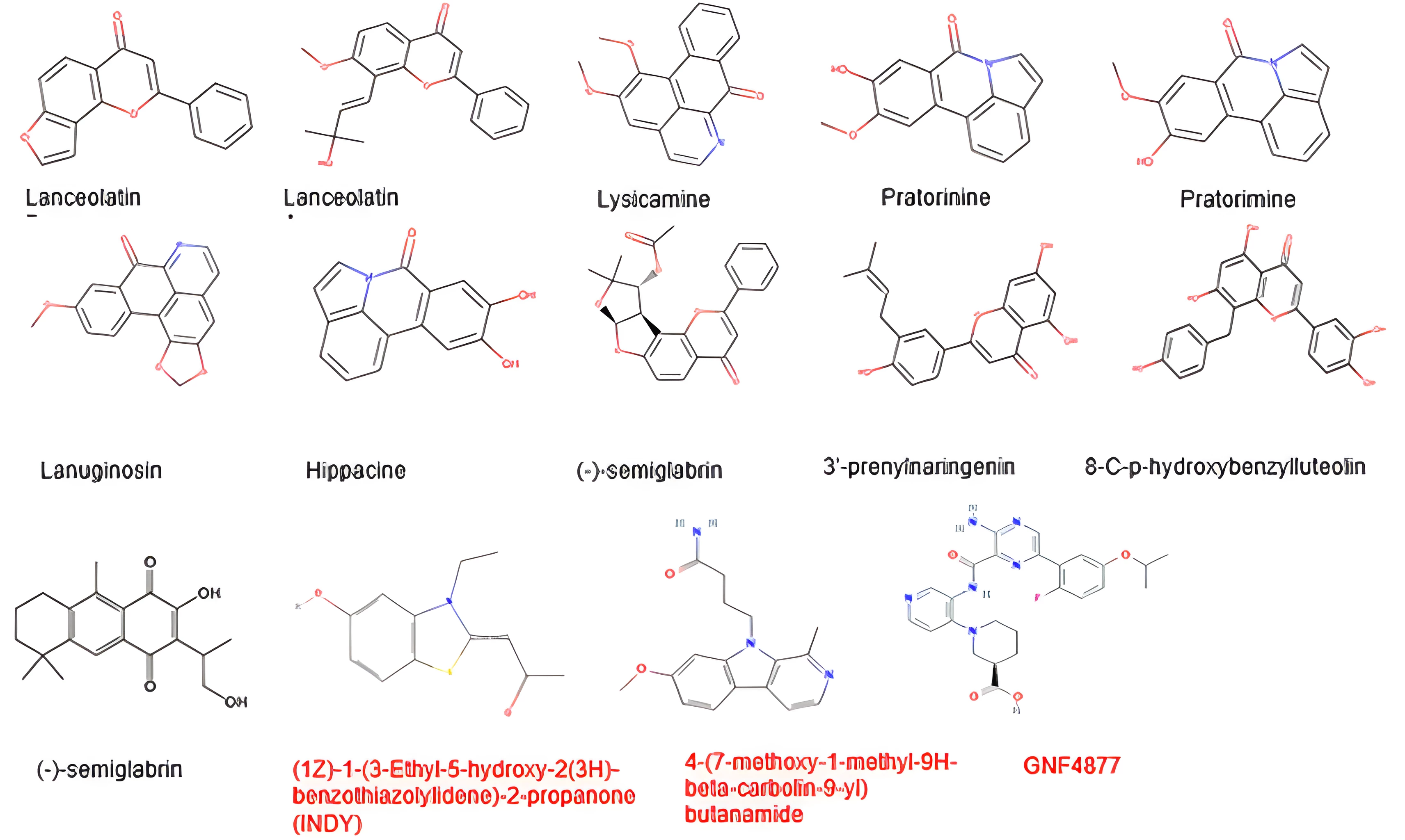
The results of in-silico studies translate well during in-vitro or in-vivo studies depending on the computational study design. In synthetic chemistry, the bioavailability, toxicity, and potential bioactivity of compound libraries are often studied with in-silico techniques before synthesis (92). Similarly, we have prepared the study design of the present in-silico study to maximize the chance of obtaining fruitful results in bioassays. Molecular docking is one of the fastest and most reliable in-silico techniques to study the binding affinity, binding pose, and molecular interactions between a ligand and a protein (93). Molecular docking studies are sometimes combined with in-vivo studies to study the molecular interaction and binding affinity of compounds with studied biomarkers (94, 95). To tackle the severity of the ongoing coronavirus disease 2019 pandemic, cancer, and other infectious diseases, several researchers have designed their in-silico studies to maximize the chance of obtaining fruitful results in bioassays (96-104). This discussion highlights the robustness of in-silico studies in drug discovery and development, corroborates and validates the study design used in the present in-silico study, and justifies that the African natural product database harbors promising phytocompounds as DYRK1A inhibitors.
Conclusions
Small-scale suppression of the DYRK1A molecules can offer a remedy for the pharmaceutical intervention of beta-cell regeneration in diabetes since options for treating beta-cell regeneration are a significant unmet therapeutic need. However, due to the conventional function of DYRK1A in controlling multiple signaling pathways vital to neuronal development and functions, caution should be used while trying to modulate it so that its activity is reduced to that which is typically seen in healthy individuals. These current in-silico tests' findings imply that 3-[6-(3-methyl-but-2-enyl)-1H-indolyl]-6-(3-methyl-but-2-enyl)-1H-indole, Lanceolatin B, Lysicamine, Pratorinine, Pratorimine, Lanceolatin A, Lanuginosine, Hippacine, (-)-Semiglabrin, Aegyptinone B,3'-Prenylnaringenin and 8-C-p-hydroxybenzylluteolin are candidate ligands for activating beta-cells regeneration. The phytocompounds exhibit good intestine absorption, according to computational analyses of drug-likeness, TPSA, and % absorption. Finally, these phytocompounds have been recognized by the in-silico analysis as prospective novel medication candidates. Validating this in-silico work requires further thorough research using different models, such as in-vivo assays using the phytocompounds or extracts containing the phytocompounds.
Declarations
Acknowledgment
We express our gratitude to the Principal Investigator, Drug Design and Informatics Group (DDIG) laboratory, for granting us free access to the facility. We appreciate the contribution of the CURIES research team of the Faculty of Pharmaceutical Sciences, Nnamdi Azikiwe University, Awka to this ongoing research.
Conflict of Interest
The authors declare no conflicting interest.
Data Availability
The unpublished data is available upon request to the corresponding author.
Ethics Statement
Not applicable.
Funding Information
Not applicable.
References
- Li Y, Teng D, Shi X, Qin G, Qin Y, Quan H, et al. Prevalence of diabetes recorded in mainland China using 2018 diagnostic criteria from the American Diabetes Association: national cross sectional study. BMJ. 2020;369:m997.
- Zhou M, Wang H, Zeng X, Yin P, Zhu J, Chen W, et al. Mortality, morbidity, and risk factors in China and its provinces, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2019;394(10204):1145-1158.
- Matveyenko AV, Butler PC. Relationship between beta-cell mass and diabetes onset. Diabetes Obes Metab. 2008;10 Suppl 4:23-31.
- Cnop M, Igoillo-Esteve M, Hughes SJ, Walker JN, Cnop I, Clark A. Longevity of human islet α- and β-cells. Diabetes Obes Metab. 2011;13 Suppl 1:39-46.
- Rieck S, Kaestner KH. Expansion of beta-cell mass in response to pregnancy. Trends Endocrinol Metab. 2010;21(4):151-158.
- Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444(7121):840-846.
- Butler AE, Cao-Minh L, Galasso R, Rizza RA, Corradin A, Cobelli C, et al. Adaptive changes in pancreatic beta-cell fractional area and beta-cell turnover in human pregnancy. Diabetologia. 2010;53(10):2167-2176.
- Mezza T, Muscogiuri G, Sorice GP, Clemente G, Hu J, Pontecorvi A, et al. Insulin resistance alters islet morphology in nondiabetic humans. Diabetes. 2014;63(3):994-1007.
- Dorrell C, Schug J, Canaday PS, Russ HA, Tarlow BD, Grompe MT, et al. Human islets contain four distinct subtypes of β cells. Nat Commun. 2016;7:11756.
- Segerstolpe Å, Palasantza A, Eliasson P, Andersson EM, Andréasson AC, Sun X, et al. Single-cell transcriptome profiling of human pancreatic islets in health and type 2 diabetes. Cell Metab. 2016;24(4):593-607.
- Li WC, Rukstalis JM, Nishimura W, Tchipashvili V, Habener JF, Sharma A, et al. Activation of pancreatic-duct-derived progenitor cells during pancreas regeneration in adult rats. J Cell Sci. 2010;123(Pt 16):2792-2802.
- Thorel F, Népote V, Avril I, Kohno K, Desgraz R, Chera S, et al. Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature. 2010;464(7292):1149-1154.
- Chera S, Baronnier D, Ghila L, Cigliola V, Jensen JN, Gu G, et al. Diabetes recovery by age-dependent conversion of pancreatic δ-cells into insulin producers. Nature. 2014;514(7523):503-507.
- Xu X, D'Hoker J, Stangé G, Bonné S, De Leu N, Xiao X, et al. Beta-cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell. 2008;132(2):197-207.
- Bonner-Weir S, Baxter LA, Schuppin GT, Smith FE. A second pathway for regeneration of adult exocrine and endocrine pancreas. A possible recapitulation of embryonic development. Diabetes. 1993;42(12):1715-1720.
- Wegiel J, Gong CX, Hwang YW. The role of DYRK1A in neurodegenerative diseases. FEBS J. 2011;278(2):236-245.
- Smith B, Medda f, Gokhale V, Dunckley T, Hulme C. Recent advances in the design, synthesis, and biological evaluation of selective DYRK1A inhibitors: a new avenue for a disease modifying treatment of Alzheimer’s? ACS Chem Neurosci. 2012;3(11):857-872.
- Ionescu A, Dufrasne F, Gelbcke M, Jabin I, Kiss R, Lamoral-Theys D. DYRK1A kinase inhibitors with emphasis on cancer. Mini Rev Med Chem. 2012;12(13):1315-1329.
- Fernandez-Martinez P, Zahonero C, Sanchez-Gomez P. DYRK1A: the double-edged kinase as a protagonist in cell growth and tumorigenesis. Mol Cell Oncol. 2015;2(1):e970048.
- Wang P, Alvarez-Perez JC, Felsenfeld DP, Liu H, Sivendran S, Bender A, et al. A high-throughput chemical screen reveals that harmine-mediated inhibition of DYRK1A increases human pancreatic beta-cell replication. Nat Med. 2015;21(4):383-388.
- Shen W, Taylor B, Jin Q, Nguyen-Tran V, Meeusen S, Zhang YQ, et al. Inhibition of DYRK1A and GSK3B induces human β-cell proliferation. Nat Commun. 2015;6:8372.
- Rachdi L, Kariyawasam D, Aiello V, Herault Y, Janel N, Delabar JM, et al. Dyrk1A induces pancreatic β cell mass expansion and improves glucose tolerance. Cell Cycle. 2014;13(14):2221-2229.
- Dirice E, Walpita D, Vetere A, Meier BC, Kahraman S, Hu J, et al. Inhibition of DYRK1A stimulates human beta-cell proliferation. Diabetes. 2016;65(6):1660-1671.
- Becker W, Soppa U, Tejedor FJ. DYRK1A: a potential drug target for multiple Down syndrome neuropathologies. CNS Neurol Disord Drug Targets. 2014;13(1):26-33.
- Guedj F, Sebrie C, Rivals I, Ledru A, Paly E, Bizot JC, et al. Green tea polyphenols rescue of brain defects induced by overexpression of DYRK1A. PLoS One. 2009;4(2):e4606.
- McLauchlan H, Elliott M, Cohen P. The specificities of protein kinase inhibitors: an update. Biochem J. 2003;371(Pt 1):199-204.
- Tahtouh T, Elkins JM, Filippakopoulos P, Soundararajan M, Burgy G, Durieu E, et al. Selectivity, cocrystal structures, and neuroprotective properties of leucettines, a family of protein kinase inhibitors derived from the marine sponge alkaloid leucettamine B. J Med Chem. 2012;55(21):9312-9330.
- Naert G, Ferre V, Meunier J, Keller E, Malmstrom S, Givalois L, et al. Leucettine L41, a DYRK1A-preferential DYRKs/CLKs inhibitor, prevents memory impairments and neurotoxicity induced by oligomeric Aβ25–35 peptide administration in mice. Eur Neuropsychopharmacol. 2015;25(11):2170-2182.
- Cozza G, Mazzorana M, Papinutto E, Bain J, Elliott M, di Maira G, et al. Quinalizarin as a potent, selective and cell-permeable inhibitor of protein kinase CK2. Biochem J. 2009;421(3):387-395.
- Ahmadu A, Abdulkarim A, Grougnet R, Myrianthopoulos V, Tillequin f, Magiatis P, et al. Two new peltogynoids from Acacia nilotica Delile with kinase inhibitory activity. Planta Med. 2010;76(5):458-460.
- Sarno S, Mazzorana M, Traynor R, Ruzzene M, Cozza G, Pagano MA, et al. Structural features underlying the selectivity of the kinase inhibitors NBC and dNBC: role of a nitro group that discriminates between CK2 and DYRK1A. Cell Mol Life Sci. 2012;69(3):449-460.
- Sanchez C, Salas AP, Brana AF, Palomino M, Pineda-Lucena A, Carbajo RJ, et al. Generation of potent and selective kinase inhibitors by combinatorial biosynthesis of glycosylated indolocarbazoles. Chem Commun (Camb). 2009;(27):4118-4120.
- Ogawa Y, Nonaka Y, Goto T, Ohnishi E, Hiramatsu T, Kii I, et al. Development of a novel selective inhibitor of the Down syndrome-related kinase Dyrk1A. Nat Commun. 2010;1:86.
- Gourdain S, Dairou J, Denhez C, Bui LC, Rodrigues-Lima f, Janel N, et al. Development of DANDYs, new 3,5-diaryl-7-azaindoles demonstrating potent DYRK1A kinase inhibitory activity. J Med Chem. 2013;56(23):9569-9585.
- Kii I, Sumida Y, Goto T, Sonamoto R, Okuno Y, Yoshida S, et al. Selective inhibition of the kinase DYRK1A by targeting its folding process. Nat Commun. 2016;7:11391.
- Koo KA, Kim ND, Chon YS, Jung MS, Lee BJ, Kim JH, et al. QSAR analysis of pyrazolidine-3,5-diones derivatives as Dyrk1A inhibitors. Bioorg Med Chem Lett. 2009;19(8):2324-2328.
- Kim ND, Yoon J, Kim JH, Lee JT, Chon YS, Hwang MK, et al. Putative therapeutic agents for the learning and memory deficits of people with Down syndrome. Bioorg Med Chem Lett. 2006;16(14):3772-3776.
- Rosenthal AS, Tanega C, Shen M, Mott BT, Bougie JM, Nguyen DT, et al. Potent and selective small molecule inhibitors of specific isoforms of Cdc2-like kinases (Clk) and dual specificity tyrosine-phosphorylation-regulated kinases (Dyrk). Bioorg Med Chem Lett. 2011;21(10):3152-3158.
- Giraud F, Alves G, Debiton E, Nauton L, Thery V, Durieu E, et al. Synthesis, protein kinase inhibitory potencies, and in vitro antiproliferative activities of meridianin derivatives. J Med Chem. 2011;54(13):4474-4489.
- Echalier A, Bettayeb K, Ferandin Y, Lozach O, Clement M, Valette A, et al. Meriolins (3-(pyrimidin-4-yl)-7-azaindoles): synthesis, kinase inhibitory activity, cellular effects, and structure of a CDK2/cyclin A/meriolin complex. J Med Chem. 2008;51(4):737-751.
- Akue-Gedu R, Debiton E, Ferandin Y, Meijer L, Prudhomme M, Anizon F. Synthesis and biological activities of aminopyrimidyl-indoles structurally related to meridianins. Bioorg Med Chem. 2009;17(13):4420-4424.
- Kassis P, Brzeszcz J, Beneteau V, Lozach O, Meijer L, Le Guevel R, et al. Synthesis and biological evaluation of new 3-(6-hydroxyindol-2-yl)-5-(phenyl) pyridine or pyrazine V-shaped molecules as kinase inhibitors and cytotoxic agents. Eur J Med Chem. 2011;46(11):5416-5434.
- Neagoie C, Vedrenne E, Buron F, Merour JY, Rosca S, Bourg S, et al. Synthesis of chromeno[3,4-b]indoles as lamellarin D analogues: a novel DYRK1A inhibitor class. Eur J Med Chem. 2012;49:379-396.
- Falke H, Chaikuad A, Becker A, Loaec N, Lozach O, Abu Jhaisha S, et al. 10-Iodo-11H-indolo[3,2-c]quinoline-6-carboxylic acids are selective inhibitors of DYRK1A. J Med Chem. 2015;58(7):3131-3143.
- Foucourt A, Hedou D, Dubouilh-Benard C, Desire L, Casagrande AS, Leblond B, et al. Design and synthesis of thiazolo[5,4-f]quinazolines as DYRK1A inhibitors, part I. Molecules. 2014;19(10):15546-15571.
- Coutadeur S, Benyamine H, Delalonde L, de Oliveira C, Leblond B, Foucourt A, et al. A novel DYRK1A (dual specificity tyrosine phosphorylation-regulated kinase 1A) inhibitor for the treatment of Alzheimer’s disease: effect on Tau and amyloid pathologies in vitro. J Neurochem. 2015;133(3):440-451.
- Abdolazimi Y, Lee S, Xu H, Allegretti P, Horton TM, Yeh B, et al. CC-401 promotes β-cell replication via pleiotropic consequences of DYRK1A/B inhibition. Endocrinology. 2018;159(9):3143-3157.
- Annes JP, Ryu JH, Lam K, Carolan PJ, Utz K, Hollister-Lock J, et al. Adenosine kinase inhibition selectively promotes rodent and porcine islet β-cell replication. Proc Natl Acad Sci U S A. 2012;109(10):3915-3920.
- Becker W, Sippl W. Activation, regulation, and inhibition of DYRK1A. FEBS J. 2011;278(2):246-256.
- Brierley DI, Davidson C. Developments in harmine pharmacology - Implications for ayahuasca use and drug-dependence treatment. Prog Neuropsychopharmacol Biol Psychiatry. 2012;39(2):263-272.
- Airaksinen MM, Lecklin A, Saano V, Tuomisto L, Gynther J. Tremorigenic effect and inhibition of tryptamine and serotonin receptor binding by β-carbolines. Pharmacol Toxicol. 1987;60(1):5-8.
- Frost D, Meechoovet B, Wang T, Gately S, Giorgetti M, Shcherbakova I, et al. β-carboline compounds, including harmine, inhibit DYRK1A and tau phosphorylation at multiple Alzheimer’s disease-related sites. PLoS One. 2011;6(5):e19264.
- Cao R, Peng W, Chen H, Ma Y, Liu X, Hou X, et al. DNA binding properties of 9-substituted harmine derivatives. Biochem Biophys Res Commun. 2005;338(3):1557-1563.
- Sobhani AM, Ebrahimi SA, Mahmoudian M. An in vitro evaluation of human DNA topoisomerase I inhibition by Peganum harmala L. seeds extract and its β-carboline alkaloids. J Pharm Pharm Sci. 2002;5(1):19-23.
- Song Y, Kesuma D, Wang J, Deng Y, Duan J, Wang JH, et al. Specific inhibition of cyclin-dependent kinases and cell proliferation by harmine. Biochem Biophys Res Commun. 2004;317(1):128-132.
- Cao MR, Li Q, Liu ZL, Liu HH, Wang W, Liao XL, et al. Harmine induces apoptosis in HepG2 cells via mitochondrial signaling pathway. Hepatobiliary Pancreat Dis Int. 2011;10(6):599-604.
- Taira Z, Kanzawa S, Dohara C, Ishida S, Matsumoto M, Sakiya Y. Intercalation of six β-carboline derivatives into DNA. Jpn J Toxicol Environ Health. 1997;43(2):83-91.
- Kim S, Chen J, Cheng T, Gindulyte A, He J, He S, et al. PubChem in 2021: new data content and improved web interfaces. Nucleic Acids Res. 2021;49(D1):D1388-D1395.
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28(1):235-242.
- Sander T, Freyss J, von Korff M, Rufener C. DataWarrior: an open-source program for chemistry aware data visualization and analysis. J Chem Inf Model. 2015;55(2):460-473.
- Schrödinger LLC. The PyMOL Molecular Graphics System, Version 1.2r3pre.
- Sanner MF. Python: a programming language for software integration and development. J Mol Graph Model. 1999;17(1):57-61.
- Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455-461.
- Molinspiration Cheminformatics. Free web services [Internet]. [cited 2021 Apr 14]. Available from: https://www.molinspiration.com.
- Simoben CV, Qaseem A, Moumbock AFA, Telukunta KK, Günther S, Sippl W, et al. Pharmacoinformatic investigation of medicinal plants from East Africa. Mol Inform. 2020;39(11):2000163.
- Ntie-Kang F, Telukunta KK, Döring K, Simoben CV, Moumbock AFA, Malange YI, et al. NANPDB: a resource for natural products from Northern African sources. J Nat Prod. 2017;80(7):2067-2076.
- Zhao YH, Abraham MH, Le J, Hersey A, Luscombe CN, Beck G, et al. Rate-limited steps of human oral absorption and QSAR studies. Pharm Res. 2002;19(10):1446-1457.
- Zothantluanga JH. Ethnopharmacology and phytochemistry-based review on the antimalarial potential of Acacia pennata (L.) Willd. Sci Vis. 2020;20(4):139-147.
- Ejiofor II, Das A, Mir SR, Ali M, Zaman K. Novel phytocompounds from Vernonia amygdalina with antimalarial potentials. Pharmacogn Res. 2020;12(1):53-59.
- Ejiofor II, Ekeomodi CC, Elomeme S, Ejiofor ME. Therapeutic inhibitors: natural product options through computer-aided drug design. In: Saxena SK, editor. Drug Repurposing - Molecular Aspects and Therapeutic Applications. IntechOpen; 2022.
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 1997;23(1-3):3-25.
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46(1-3):3-26.
- Lipinski CA. Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov Today Technol. 2004;1(4):337-341.
- Accelrys. BIOVIA Databases | Bioactivity Databases: RTECS [Internet]. [cited 2021 Apr 15]. Available from: accelrys.com.
- Sergeev YV, Dolinska MB, Wingfield PT. Thermodynamic analysis of weak protein interactions using sedimentation equilibrium. Curr Protoc Protein Sci. 2014;77:20.12.1-20.12.15.
- Du X, Li Y, Xia YL, Ai SM, Liang J, Sang P, et al. Insights into protein-ligand interactions: mechanisms, models, and methods. Int J Mol Sci. 2016;17(2):144.
- Muthu S, Durairaj B. Molecular docking studies on interaction of Annona muricata compounds with antiapoptotic proteins Bcl-2 and survivin. Sky J Biochem Res. 2016;5(2):14-17.
- Clark DE. Rapid calculation of polar molecular surface area and its application to the prediction of transport phenomena. 2. Prediction of blood-brain barrier penetration. J Pharm Sci. 1999;88(8):815-821.
- Kelder J, Grootenhuis PD, Bayada DM, Delbressine LP, Ploemen JP. Polar molecular surface as a dominating determinant for oral absorption and brain penetration of drugs. Pharm Res. 1999;16(10):1514-1519.
- De la Torre R, De Sola S, Pons M, Duchon A, de Lagran MM, Farré M, et al. Epigallocatechin-3-gallate, a DYRK1A inhibitor, rescues cognitive deficits in Down syndrome mouse models and in humans. Mol Nutr Food Res. 2014;58(2):278-288.
- Royal Botanic Gardens, Kew. Acacia nilotica (acacia) | Plants & Fungi At Kew [Internet]. [cited 2021 Jun 17]. Available from: www.kew.org/science-conservation/plants-fungi.
- Stotani S, Giordanetto F, Medda F. DYRK1A inhibition as potential treatment for Alzheimer’s disease. Future Med Chem. 2016;8(6):681-696.
- Kelland LR. Flavopiridol, the first cyclin-dependent kinase inhibitor to enter the clinic: current status. Expert Opin Investig Drugs. 2000;9(12):2903-2911.
- Raje N, Hideshima T, Mukherjee S, Raab M, Vallet S, Chhetri S, et al. Preclinical activity of P276-00, a novel small-molecule cyclin-dependent kinase inhibitor in the therapy of multiple myeloma. Leukemia. 2009;23(5):961-970.
- Nguyen TB, Lozach O, Surpateanu G, Wang Q, Retailleau P, Iorga BI, et al. Synthesis, biological evaluation, and molecular modeling of natural and unnatural flavonoidal alkaloids, inhibitors of kinases. J Med Chem. 2012;55(6):2811-2819.
- Omura S, Iwai Y, Hirano A, Nakagawa A, Awaya J, Tsuchya H, et al. A new alkaloid AM-2282 of Streptomyces origin. Taxonomy, fermentation, isolation and preliminary characterization. J Antibiot (Tokyo). 1977;30(4):275-282.
- Rüegg UT, Burgess GM. Staurosporine, K-252 and UCN-01: potent but nonspecific inhibitors of protein kinases. Trends Pharmacol Sci. 1989;10(6):218-220.
- Sánchez C, Salas AP, Braña AF, Palomino M, Pineda-Lucena A, Carbajo RJ, et al. Generation of potent and selective kinase inhibitors by combinatorial biosynthesis of glycosylated indolocarbazoles. Chem Commun (Camb). 2009;(27):4118-4120.
- Beniddir MA, Le Borgne E, Iorga BI, Loaëc N, Lozach O, Meijer L, et al. Acridone alkaloids from Glycosmis chlorosperma as DYRK1A inhibitors. J Nat Prod. 2014;77(5):1117-1122.
- Deslandes S, Chassaing S, Delfourne E. Synthesis of two series of pyrazolic analogs of the marine alkaloids granulatimide and isogranulatimide as potent checkpoint 1 kinase inhibitors. Tetrahedron Lett. 2010;51(43):5640-5642.
- Deslandes S, Lamoral-Theys D, Frongia C, Chassaing S, Bruyère C, Lozach O, et al. Synthesis and biological evaluation of analogs of the marine alkaloids granulatimide and isogranulatimide. Eur J Med Chem. 2012;54:626-636.
- Kalita J, Chetia D, Rudrapal M. Design, synthesis, antimalarial activity and docking study of 7-chloro-4-(2-(substituted benzylidene)hydrazineyl)quinolones. Med Chem. 2022;18(7):928-937.
- Zothantluanga JH. Molecular docking simulation studies, toxicity study, bioactivity prediction, and structure-activity relationship reveals rutin as a potential inhibitor of SARS-CoV-2 3CL pro. J Sci Res. 2021;65(5):96-104.
- Pasala PK, Shaik RA, Rudrapal M, Khan J, Alaidarous MA, Khairnar SJ, et al. Cerebroprotective effect of Aloe emodin: in silico and in vivo studies. Saudi J Biol Sci. 2022;29(2):998-1005.
- Pasala PK, Uppara RK, Rudrapal M, Zothantluanga JH, Umar AK. Silybin phytosome attenuates cerebral ischemia-reperfusion injury in rats by suppressing oxidative stress and reducing inflammatory response: in vivo and in silico approaches. J Biochem Mol Toxicol. 2022;36(7):e23073.
- Rudrapal M, Celik I, Khan J, Ansari MA, Alomary MN, Alatawi FA, et al. Identification of bioactive molecules from Triphala (Ayurvedic herbal formulation) as potential inhibitors of SARS-CoV-2 main protease (Mpro) through computational investigations. J King Saud Univ Sci. 2022;34(3):101826.
- Zothantluanga JH, Gogoi N, Shakya A, Chetia D, Lalthanzara H. Computational guided identification of potential leads from Acacia pennata (L.) Willd. as inhibitors for cellular entry and viral replication of SARS-CoV-2. Future J Pharm Sci. 2021;7(1):201.
- Umar AK, Zothantluanga JH. Structure-based virtual screening and molecular dynamics of quercetin and its natural derivatives as potent oxidative stress modulators in ROS-induced cancer. Indones J Pharm. 2021;32(1):60-72.
- Patowary L, Borthakur MS, Zothantluanga JH, Chetia D. Repurposing of FDA approved drugs having structural similarity to artemisinin against PfDHFR-TS through molecular docking and molecular dynamics simulation studies. Curr Trends Pharm Sci. 2021;8(2):14-34.
- Umar AK, Kelutur FJ, Zothantluanga JH. Flavonoid compounds of buah merah (Pandanus conoideus Lamk) as a potent oxidative stress modulator in ROS-induced cancer: in silico approach. Maj Obat Tradis. 2022;26(3):221-232.
- Rudrapal M, Celik I, Chinnam S, Ansari MA, Khan J, Alghamdi S, et al. Phytocompounds as potential inhibitors of SARS-CoV-2 Mpro and PLpro through computational studies. Saudi J Biol Sci. 2022;29(5):3456-3465.
- Zothantluanga JH, Abdalla M, Rudrapal M, Tian Q, Chetia D, Li J. Computational investigations for identification of bioactive molecules from Baccaurea ramiflora and Bergenia ciliata as inhibitors of SARS-CoV-2 Mpro. Polycycl Aromat Compd. 2022;1-29. doi: 10.1080/10406638.2022.2046613.
- Umar AK, Zothantluanga JH, Aswin K, Maulana S, Zubair MS, Lalhlenmawia H, et al. Antiviral phytocompounds “ellagic acid” and “(+)-sesamin” of Bridelia retusa identified as potential inhibitors of SARS-CoV-2 3CL pro using extensive molecular docking, molecular dynamics simulation studies, binding free energy calculations, and bioactivity prediction. Struct Chem. 2022;33(5):1473-1490.
- Zothantluanga JH, Aswin K, Rudrapal M, Chetia D. Antimalarial flavonoid-glycoside from Acacia pennata with inhibitory potential against PfDHFR-TS: an in-silico study. Biointerface Res Appl Chem. 2022;12(4):4871-4887.