
RESEARCH ARTICLE
Computational Studies of Bridelia Retusa Phytochemicals for the Identification of Promising Molecules with Inhibitory Potential Against the Spike Protein and Papain-Like Protease of SARS-CoV-2
Sciences of Phytochemistry|Vol. 1, Issue 1, pp. 26-35 (2022)
CC BY 4.0-2022 Authors
Views
Downloads
Shares
Received
Jun 28, 2022Revised
Jul 5, 2022Accepted
Jul 5, 2022Published
Jul 6, 2022
Abstract
SARS-CoV-2 is the pathogen responsible for the on-going COVID-19 pandemic. The two proteins namely, spike protein and papain-like protease are mainly responsible for the penetration and transmission of the virus, respectively. The objective of our study was to find the most promising phytoconstituents of Bridelia retusa that can inhibit both the proteins. Molecular docking, protein-ligand interactions, and molecular dynamics (MD) simulation techniques were used in the study. Bepridil and the co-crystal inhibitors of each protein were used as the standards. All the 14 phytoconstituents along with the standard drug and the co-crystal inhibitor of each protein were subjected to molecular docking. Ten compounds showed better binding affinities than the standards against the spike protein and 7 compounds have shown better binding affinities than the standards against papain-like protease protein. From the protein-ligand interactions, a total of 3 out of 10 for the spike protein and 5 out of 7 for the papain-like protease showed better interactions than the standards. An all-atom MD simulations study revealed that (R)4-(1,5-dimethyl-3-oxo-4-hexenyl)-benzoic acid formed the most stable complex with both proteins. The in-silico study provides an evidence for (R)4-(1,5-dimethyl-3-oxo-4-hexenyl)-benzoic acid as a promising inhibitor of the spike and papain-like protease of SARS-CoV-2. Further investigations such as in-vitro/in-vivo studies are recommended to validate the potency of (R)4-(1,5-dimethyl-3-oxo-4-hexenyl)-benzoic acid.
Keywords:
Introduction
Severe Acute Respiratory Syndrome (SARS), a viral respiratory illness, is caused by a coronavirus called SARS-associated coronavirus (SARS-CoV). SARS-CoV-2 is the virus responsible for the dreadful infectious coronavirus disease 2019 (COVID-19). The symptoms of COVID-19 are fever, cough, tiredness, loss of taste and smell, headache, sore throat, aches & pains, diarrhea, difficulty in breathing, and chest pain. Majority of the infected people who developed mild to moderate symptoms can recover without hospitalization whereas medical attention is required in case of serious symptoms (1). By the end of 2019, a novel coronavirus designated as SARS-CoV-2 emerged in the city of Wuhan, China, and made an outbreak of unusual viral pneumonia (2).
To date, a total of 539,119,771 confirmed cases of COVID-19, of which 6,322,311 deaths have been reported by the World Health Organisation (WHO) (3). The SARS-CoV-2 is a member of the beta coronavirus (beta-CoV) family, which includes RNA viruses with spikes resembling crowns on the surface of the coronavirus particles, similar to the SARS (severe acute respiratory syndrome) and MERS (Middle East Respiratory Syndrome) viruses. However, the WHO estimates that the death rate for SARS-CoV-2 is between 2 and 3 percent, compared to 14% and 35% for SARS and MERS, respectively. However, the SARS-CoV-2 possesses characteristics such as rapid person-to-person transmission, asymptomatic transmission, protracted symptom development, as well as significantly higher fatality rates in the older population (4).
The pandemic has increased the demand for critical care, placing enormous strain on the healthcare systems of many nations. Many therapies were suggested for the treatment of SARS-CoV-2 during the peak COVID-19 pandemic. The WHO has launched a trial, SOLIDARITY, to focus on testing the four most promising COVID-19 treatments namely, remdesivir, chloroquine and hydroxy-chloroquine, lopinavir, and lopinavir and ritonavir and interferon-beta. Chloroquine/hydroxy-chloroquine and lopinavir/ritonavir were removed from the COVID-19 treatment protocols in June 2020 due to possible risks and uncertainty of their benefits. Remdesivir was the first and only drug approved by the FDA on 22 October 2020 (5, 6).
However, SARS-CoV-2 kept mutating leading to the emergence of deadlier variants with higher transmissibility which might weaken the efficacy of the available vaccines and antiviral medications. Therefore, the search for novel or adjuvant anti-SARS-CoV-2 medications is urgently required. Investigating the bioactive compounds of plants appears to be a promising approach for the identification and development of new medications for COVID-19 (7). Numerous plants have been reported to possess antiviral phytoconstituents that may be beneficial against SARS-CoV-2 namely, Lycoris radiata, Artemisia annua, Pyrrosia lingaua, Lindera aggregate, Isatis indigotica, Torreya nucifera, Houttuynia cordata, Curcuma longa, and Curcuma xanthorrhiza (8, 9).
Bridelia retusa (L. ) A. Juss. is a deciduous tree found throughout India that grows to be about 20 meters tall. The plant is used as a traditional herbal remedy. Natives use the stem bark and roots to treat rheumatism and as astringent agents (10). During fever, a powdered stem bark mixed with water is administered (11). The plant is said to have a variety of biological activities such as anti-viral, respiratory, cardiovascular, and anticancer activities (12). In-vivo studies have also revealed that B. retusa has antinociceptive, anti-inflammatory, antimicrobial, and anti-fungal properties and also stimulates cell-mediated immunity (13-15). The facts presented above supports the rationale for investigating B. retusa phytocompounds as a potential antiviral agent against SARS-CoV-2. Phytoconstituents like tannins, triterpene ketone, decanoic acid octadecyl ester, stigmasterol, and dehydrostigmasterol are found in the bark. The fruits contain β-sitosterol, gallic acid, and ellagic acid and the leaves contain crude proteins (16).
In our present study, we have selected 14 phytocompounds namely lupeol, friedelin, gallic acid, ellagic acid, (E)-4-(1, 5-dimethyl-3-oxo-1, 4-hexadienyl) benzoic acid, (E)-4-(1, 5-dimethyl-3-oxo-1-hexenyl) benzoic acid, (R)-4-(1, 5-dimethyl-3-oxo-4-hexenyl) benzoic acid, (-)-isochaminic acid, (R)-4-(1, 5-dimethyl-3-oxohexyl) benzoic acid (ar-todomatuic acid), 5-allyl-1, 2, 3-trimethoxybenzene (elemicin), (+)-sesamin, β-sitosterol, stigmasterol, and 4-isopropylbenzoic acid (cumic acid) that have been reported to be present in B. retusa and were investigated for their inhibitory potential against the main protease of SARS-CoV-2 through in silico approach (17). In SARS-CoV-2, the spike protein (S) help the virus to penetrate the host cell (18) and the papain-like protease (PL pro) protein is necessary for the processing of viral polyproteins to generate a functional replicase complex and enable the spread of the virus (19). In the present study, the inhibitory potential of the selected 14 phytocompounds against these protein targets will be investigated with in silico techniques.
Materials and Methods
Retrieval of Target Protein
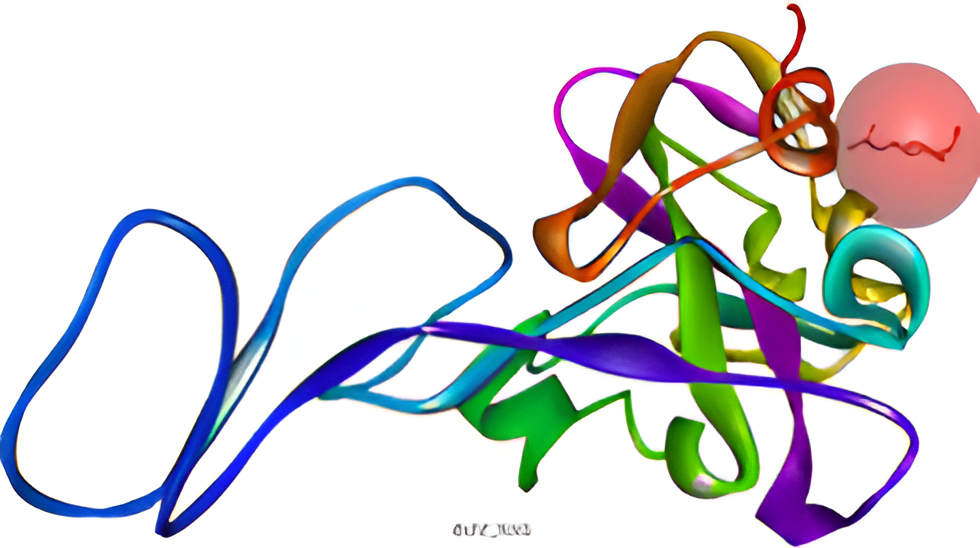
The X-ray crystal structure of SARS-CoV-2 S-protein (PDB ID: 6M0J) and PL pro (PDB ID: 7OFT) are selected as the target proteins. They were downloaded in ‘. pdb’ format from the RCSB-PDB website. The crystal structures of the target chains of the proteins are shown in Figure 1 and Figure 2. The S-protein was found to have two Chains (Chain A and Chain E) and the PL pro has only one Chain (Chain A). The co-crystal inhibitor of the S-protein (2-acetamido-2-deoxy-beta-D-glucopyranose, NAG) and PL pro protein (p-hydroxybenzaldehyde, HBA) was identified and they manually prepared with the Chem Draw Professional 16.0.0.82 [68] software.


Preparation of Target Proteins
The target proteins were prepared with the BIOVIA Discovery studio Visualizer v21.1.0.20298 software (20). The Chain E (S-protein) and Chain A (PL pro) were used for the study and the other chains were deleted from the target proteins. The water molecules and heteroatoms were also removed from the target proteins. Now the polar hydrogen was added to both the target proteins. Then the active binding sites were defined with the ‘define and edit binding site’ feature of Discovery Studio Software and active site coordinates of S-protein (x=-35.41, y=8.55, z=29.13) and PL pro (x= 36.56, y= 9.35, z= 16.36) were saved for future use. The prepared proteins were saved in the PDB file format for future use.
Preparation of Compound Library
The structure of the fourteen phytoconstituents of B. retusa was prepared manually with the Chem Draw Professional 16.0.0.82[68] software. These constituents were already reported as potent against SARS-CoV-2 3CLPro protein (17). Further, the SMILES ID of the structures was also saved for future use. The prepared compounds were saved in MDL SD File (*SDF) format. The structure of the standard drug Bepridil has been obtained from the PubChem database (PubChem CID 2351) and it was saved in the *SDF format.
Molecular Docking
The molecular docking simulation study (MDSS) was carried out with Autodock Vina on the virtual screening tool PyRx 0.8 software (21). The prepared proteins were loaded in the 3D scene in the virtual platform of the software and converted to the PDBQT file format when made into macromolecules (22). The originally downloaded proteins were reloaded and the unnecessary chains were removed from the scene except for Chain E (S-protein) and Chain A (PL pro). The sequence of the amino acids and the co-crystal ligand were revealed on expanding Chain E and Chain A of the respective proteins. The atoms of the co-crystal ligands were labeled to determine the accurate location of the co-crystal inhibitors present at the binding site of the protein. In the Vina search space of the PyRx 0.8 tool, the pre-defined active binding site coordinates were used to adjust the alignment of the 3D affinity grid box so that all the amino acids get covered at the active binding site of the protein. The size of the 3D affinity grid box was kept at default ay 25 Å. Finally, according to the standard protocols of the PyRx tool, MDSS were carried out (23).
Visualisation and Analysis of Ligand Interactions
The 2D interaction of the phytoconstituents with the best binding affinities with both the proteins was visualized with the Discovery Studio Visualizer Software. The 2D ligand interactions of the co-crystal-proteins was also visualized. The compound that did not form any conventional hydrogen bond with the active site residues was discarded from the study.
Molecular Dynamics Simulations
The molecular dynamics (MD) simulations studies was used to predict the most stable protein-ligand complex based on the vales of root mean square fluctuation (RMSF). A protein-ligand complex is considered to be stable if the RMSF value of all the amino acids is lower than 2.0Å (24, 25). A protein-ligand complex of each constituent with S-protein and PL pro were generated and saved in the PDB file format. The MD simulations was carried out for only proteins along with the prepared complexes. The MD simulations was performed on the CABS Flex 2.0 server which utilizes the coarse-grained simulations of the protein motion (26). All the parameters of the MD simulation study were kept at default.
Results
Features of Target Proteins
The crystal structure of the chain E of ‘S’ protein and chain A of ‘PL Pro’ protein were retrieved from the RCSB-PDB website, shown in Figure 3 and Figure 4 respectively. The S-protein is made up of two chains, i. e, Chain A (sequence length 603), and Chain E (sequence length 229). The Chain E of the protein is complexed with co-crystal inhibitor viz. NAG (2-acetamido-2-deoxy-beta-D-glucopyranose). The PL pro is composed of one chain, i. e, Chain A (sequence length 315), complexed with HBA (p-hydroxy benzaldehyde).

Molecular Docking and Visualization of Ligand Interactions
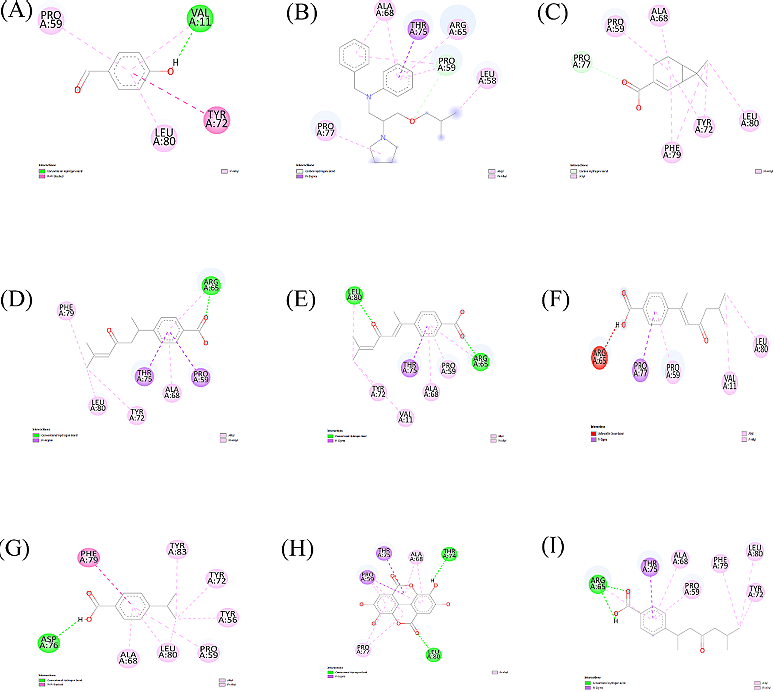
MDSS is a reliable tool for in silico drug screening. PyRx docking provides binding affinity values (-kcal/mol) to rank ligand-protein interactions (Table 1). Each ligand generates up to nine binding poses. Ligand interactions were visualized using Discovery Studio Visualizer, with 2D images shown in Figures 5 and 6. Tables 2 and 3 summarize ligand-amino acid interactions.
| Drug | Binding affinity (-kcal/mol)S-protein | Binding affinity (-kcal/mol)PL pro |
|---|---|---|
| Co-crystal inhibitor | 4.8 | 6.2 |
| Bepridil (Standard) | 6 | 7.2 |
| (-)-Isochaminic acid | 5.3 | 7.6 |
| (+)- Sesamin | 8.5 | 6.2 |
| β-Sitosterol | 7.7 | 6.7 |
| Lupeol | 7.7 | 6.2 |
| (R)4-(1,5-dimethyl-3-oxo-4-hexenyl)-benzoic acid | 6.4 | 8.2 |
| (E)4-(1,5-dimethyl-3-oxo-1-hexenyl)-benzoic acid | 6.5 | 7.8 |
| (E)4-(1,5-dimethyl-3-oxo-1,4-hexadienyl)-benzoic acid | 6.7 | 8.4 |
| Elemicin | 5.3 | 5.7 |
| Stigmasterol | 8.1 | 6.7 |
| Gallic acid | 4.7 | 6.7 |
| Friedelin | 8.6 | 6.6 |
| Cumic acid | 5.5 | 7.6 |
| Ellagic acid | 6.3 | 8.3 |
| Ar-Todomatuic acid | 6.1 | 7.8 |
| Drugs | Conventional Hydrogen bond | Other interactive sites |
|---|---|---|
| NAG (Co-crystal inhibitor) | ASN343 (2.61Å) | LEU368 (3.73Å) |
| Bepridil | -- | PHE342 (4.41 Å), LEU368 (4.99 Å), SER371 (3.71 Å), TRP436 (5.02 Å) |
| (+)- Sesamin | -- | VAL367 (3.58Å), PHE374 (4.70Å), TRP436 (5.26Å, 5.50Å) |
| (E)-4-(1,5-dimethyl-3-oxo-1,4-hexadienyl) benzoic acid | -- | PHE342 (3.88Å), VAL367 (4.11Å), TRP436 (6.85Å) |
| (E)-4-(1,5-dimethyl-3-oxo-1-hexenyl) benzoic acid | -- | VAL367, SER373, TRP436 |
| (R)-4-(1,5-dimethyl-3-oxo-4-hexenyl) benzoic acid | CYS336 (2.00Å), ASN343 (2.59Å), ASP364 (1.83Å) | GLY339 (3.64Å), PHE342 (4.12Å), ALA363 (3.50Å), LEU368 (3.95Å), PHE374 (4.99Å) |
| Ar-todomatuic acid | ASN343 (2.41Å), VAL362 (2.71Å), ASP364 (1.76Å) | PHE342 (3.93Å), LEU368 (3.88Å), PHE374 (4.93Å) |
| β-Sitosterol | PHE342 (4.91Å), LEU368 (4.80Å), PHE374 (5.24Å) | |
| Ellagic acid | CYS336 (2.07Å), GLY339 (2.22Å) | VAL367 (3.64Å, 4.64Å), LEU368 (5.25Å, 5.25Å) |
| Stigmasterol | -- | PHE342 (5.11Å), LEU368 (5.08Å), PHE374 (5.10Å), TRP436 (3.89 Å, 4.62Å) |

| Drugs | Conventional Hydrogen bond | Other interactive sites |
|---|---|---|
| HBA (Co-crystal inhibitor) | VAL11 (2.05Å) | VAL11 (5.17Å), PRO59 (5.40Å), TYR72 (4.86Å), LEU80 (4.09Å) |
| Bepridil | -- | PRO77 (4.14Å), ALA68 (4.37Å, 4.40 Å), THR75 (3.65Å), ARG65 (4.47Å), PRO59 (4.50Å, 5.06Å, 3.44Å), LEU58 (4.36Å) |
| (-)-Isochaminic acid | -- | PRO77 (4.46Å), PRO59 (5.29Å), ALA68 (4.48Å), PHE79 (5.09Å, 5.17Å), TYR72 (5.40Å, 4.07Å), LEU80 (4.21Å) |
| (R)4-(1,5-dimethyl-3-oxo-4-hexenyl)-benzoic acid | ARG65 (1.91Å) | ARG65 (4.84Å), PHE79 (5.15Å), PRO59 (3.98Å), LEU80 (4.10Å), ALA68 (4.25Å), TYR72 (4.21Å), THR75 (3.81Å) |
| (E)4-(1,5-dimethyl-3-oxo-1-hexenyl)-benzoic acid | -- | PRO77 (3.73Å), PRO59 (4.00Å), VAL11 (4.19Å), LEU80 (3.76Å) |
| (E)4-(1,5-dimethyl-3-oxo-1,4-hexadienyl)-benzoic acid | ARG65 (1.92Å), LEU80 (2.04Å) | ARG65 (4.41Å), LEU80 (4.19Å), PRO59 (4.41Å), ALA68 (4.47Å), THR75 (3.74Å), VAL11 (4.97Å), TYR72 (4.26Å) |
| Cumic acid | ASP76 (1.98Å) | PHE79 (5.40Å), ALA68 (4.41Å), LEU80 (5.27Å, 5.03Å), PRO59 (4.20Å), TYR56 (5.20Å), TYR72 (4.19Å), TYR83 (4.72Å) |
| Ellagic acid | LEU80 (1.98Å), THR74 (2.45Å) | ALA68 (4.12Å, 4.20Å), THR75 (3.83Å), PRO59 (3.57Å, 4.81Å, 3.76Å, 3.93Å), PRO77 (5.23Å, 5.45Å) |
| Ar-Todomatuic acid | ARG65 (1.96Å, 2.02Å) | ARG65 (4.62Å), THR75 (3.72Å), ALA68 (4.31Å), PRO59 (4.42Å), PHE79 (4.89Å), LEU80 (4.33Å), TYR 72 (4.01Å) |
Molecular Dynamic Simulation
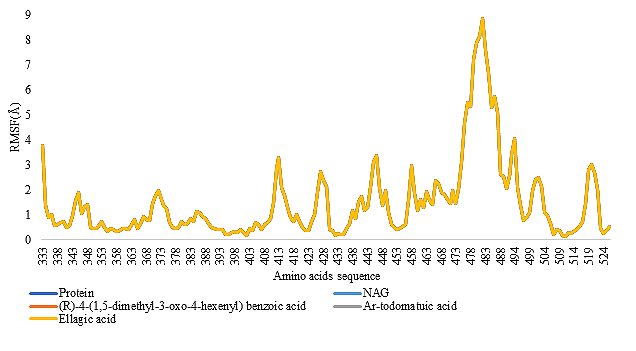
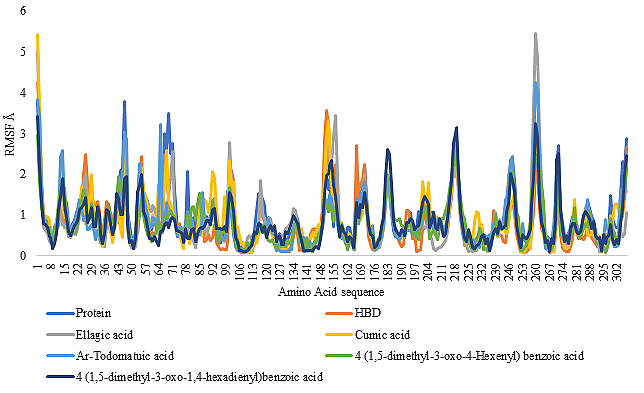
The MD simulation studies performed in the present study generated the RMSF values for each amino acid residue of the protein thereby furnishing an idea on the stability of each amino acid under a given set of parameters for a period of 10 nanoseconds. This study helps to validate the conformational stability of the protein-ligand complexes. The RMSF plot of the amino acids of S-protein and PL pro, both alone and in complex with all the phytoconstituents are given in Figure 7 and Figure 8 respectively.
Discussion
MDSS is a computational technique for searching for an appropriate ligand that fits both energetically and geometrically the binding site of a protein (27). A more negative binding affinity value suggests a better binding between a compound and a protein (23). A low binding affinity value also indicates the low energy requirement for protein-ligand binding (27). In all cases, the first pose is considered the best pose since it has the highest binding affinity and the last pose shows the lowest binding affinity towards the target protein.
Of all the compounds, 10 phytoconstituents of B. retusa have shown better binding affinity towards Chain E of the S-protein as compared to the binding affinities of the standards. These 10 constituents include (+)-Sesamin (-8.5 kcal/mol), (R)4-(1, 5-dimethyl-3-oxo-4-hexenyl)-benzoic acid (-6.4 kcal/mol), (E)4-(1, 5-dimethyl-3-oxo-1-hexenyl)-benzoic acid (-6.5 kcal/mol), (E)4-(1, 5-dimethyl-3-oxo-1, 4-hexadienyl)-benzoic acid (-6.7 kcal/mol), Stigmasterol (-8.1 kcal/mol), Friedelin (-8.6 kcal/mol), Ellagic acid (-6.3 kcal/mol), Ar-Todomatuic acid (-6.1 kcal/mol). Towards Chain A of PL pro, 7 phytoconstituents have shown better binding affinity than the standards. These include (-)-Isochaminic acid (-7.6 kcal/mol), (R)4-(1, 5-dimethyl-3-oxo-4-hexenyl)-benzoic acid (-8.2 kcal/mol), (E)4-(1, 5-dimethyl-3-oxo-1-hexenyl)-benzoic acid (-7.8 kcal/mol), (E)4-(1, 5-dimethyl-3-oxo-1, 4-hexadienyl)-benzoic acid (-8.4kcal/mol), Cumic acid (-7.6 kcal/mol), Ellagic acid (-8.3 kcal/mol), Ar-Todomatuic acid (-7.8 kcal/mol). These phytoconstituents are taken for visualization of ligand interactions and MD simulations study.
From the ligand interactions in Figures 5, 6, and Tables 2, and 3 above, it can be observed that for the S-protein, only four out of a total of sixteen compounds (including standards) has formed a conventional hydrogen bond with different amino acids at the active site. Bepridil,(+)- sesamin, (E)-4-(1,5-dimethyl-3-oxo-1,4-hexadienyl) benzoic acid, (E)-4-(1,5-dimethyl-3-oxo-1-hexenyl) benzoic acid, β-Sitosterol, stigmasterol were not able to form hydrogen bond. Therefore, the remaining four compounds were taken for further analysis. Similarly, for the PL pro, most of the phytoconstituents have formed a conventional hydrogen bond with different amino acids of the active site. The three compounds namely, Bepridil,(-)-isochaminic acid, and (E)4-(1,5-dimethyl-3-oxo-1-hexenyl)-benzoic acid were not able to form any hydrogen bond. Therefore, the remaining six phytoconstituents were taken for the MD simulations study.
For each amino acid, a lower RMSF value indicates limited flexibility and a higher RMSF value indicates high flexibility in a given system (25). For the S-protein, the RMSF value for 78.75% of all the amino acids of the protein without the presence of any ligand was found to be below 2 Å. For the S-NAG complex, the RMSF value of a total of 78.75% amino acids was found to be lower than 2Å. For the S-(R)-4-(1,5-dimethyl-3-oxo-4-hexenyl) benzoic acid complex, S-Ar-todomatuic acid complex, S-Ellagic acid complex, 78.75% amino acids have exhibited RMSF value lower than 2Å. For the PL Pro protein, without the presence of any ligand, the RMSF value of a total of 92.18% amino acids was found to be lower than 2Å. For PL Pro-HBD complex, the RMSF value of a total of 92.51% amino acids was found to be lower than 2Å. For PL Pro-Ellagic acid complex, the RMSF value of a total of 93.16% amino acids was found to be lower than 2 Å. For PL Pro-Cumic acid, the RMSF value of a total of 94.46% amino acids was found to be lower than 2Å. For PL Pro-Ar-todomatuic acid complex, the RMSF value of a total of 93.49% amino acids was found to be lower than 2Å.
For PL Pro- (R)4-(1,5-dimethyl-3-oxo-4-hexenyl)-benzoic acid complex, the RMSF value of a total of 97.07% amino acids was found to be lower than 2 Å. For PL Pro-(E)4-(1,5-dimethyl-3-oxo-1,4-hexadienyl)-benzoic acid complex, the RMSF value of a total of 94.46% amino acids was found to be lower than 2 Å. The constituents of B. retusa namely, (R)4-(1,5-dimethyl-3-oxo-4-hexenyl)-benzoic acid, Ellagic acid, Ar-Todomatuic acid were able to form the same stable complexes similar to that of its co-crystal inhibitors of the S-protein. Phytoconstituents such as cumic acid, E-4-(1,5-dimethyl-3-oxo-1,4-hexadienyl)-benzoic acid has been able to form a more stable complex with the PL pro than that of its co-crystal inhibitor. Of these (R)4-(1,5-dimethyl-3-oxo-4-hexenyl)-benzoic acid is found to form the most stable complex with PL pro.
Nowadays, researchers have used new research methods that deviates from the traditional herbal research. For example, in silico techniques such as MDSS and MD simulations have been increasingly applied in drug discovery research to identify promising phytocompounds for the treatment of various diseases (17, 28-39). Medicinal plants are an important source of clinically important phytocompounds and many pharmaceutical drugs have been developed from traditional herbal remedies (40-44). However, some phytocompounds have poor oral bioavailability. To overcome the bioavailability issues associated with natural products, novel drug delivery systems have been adopted by many researchers as the solution (45, 46). Artificial intelligence, machine learning, and supervised machine learning have also been utilized in the process of drug discovery and development (47, 48). With the advancement in science, pharmaceutical researchers are using new techniques in drug discovery programs. In the present study, we have also utilized in silico techniques that is sustainable, safe, and economical for the identification of promising bioactive molecules against SARS-CoV-2.
Conclusions
The present in silico study has revealed that the phytoconstituent (R)4-(1,5-dimethyl-3-oxo-4-hexenyl)-benzoic acid of B. retusa is a potential inhibitor of the S-protein and PL pro of the SARS-CoV-2. It has exhibited better binding affinity towards the active binding site of the S-protein and PL pro in comparison to the standards. (R)4-(1,5-dimethyl-3-oxo-4-hexenyl)-benzoic acid has shown similar molecular interactions with that of the standards in the active binding sites of both the proteins. However, the present study is confined to only the in silico model, hence, further, (in vitro/in vivo) study is needed to determine the complete inhibitory potential of the phytocompound against S-protein and PL pro of SARS-CoV-2.
Declarations
Conflict of Interest
The authors declare no conflicting interest.
Data Availability
The unpublished data is available upon request to the corresponding author.
Ethics Statement
Not applicable.
Funding Information
Not applicable.
References
- World Health Organization. WHO Coronavirus [Internet]. Geneva: World Health Organization; 2022 [cited 2022 Jun 26]. Available from: https://www.who.int/health-topics/coronavirus
- Centers for Disease Control and Prevention. CDC Coronavirus [Internet]. Atlanta: CDC; 2022 [cited 2022 Jun 26]. Available from: https://www.cdc.gov/coronavirus/2019-ncov/index.html
- World Health Organization. WHO Coronavirus (COVID-19) Dashboard [Internet]. Geneva: World Health Organization; 2022 [cited 2022 Jun 26]. Available from: https://covid19.who.int/
- Hosseini M, Chen W, Xiao D, Wang C. Computational molecular docking and virtual screening revealed promising SARS-CoV-2 drugs. Precis Clin Med. 2021;4(1):1-16.
- Bhimraj A, Morgan RL, Shumaker AH, Lavergne V, Baden L, Cheng VCC, et al. Infectious Diseases Society of America Guidelines on the treatment and management of patients with coronavirus disease 2019 (COVID-19). Clin Infect Dis. 2020;ciaa478. doi: 10.1093/cid/ciaa478
- US Food and Drug Administration. FDA Approves First Treatment for COVID-19 [Internet]. Silver Spring: FDA; 2022 [cited 2022 Jun 26]. Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-covid-19
- ul Qamar MT, Alqahtani SM, Alamri MA, Chen LL. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J Pharm Anal. 2020;10(4):313-319.
- Panyod S, Ho CT, Sheen LY. Dietary therapy and herbal medicine for COVID-19 prevention: A review and perspective. J Tradit Complement Med. 2020;10(4):420-427.
- Nugraha RV, Ridwansyah H, Ghozali M, Khairani AF, Atik N. Traditional herbal medicine candidates as complementary treatments for COVID-19: a review of their mechanisms, pros and cons. Evid Based Complement Alternat Med. 2020;2020:2560645. doi: 10.1155/2020/2560645
- Jayasinghe L, Kumarihamy BM, Jayarathna KN, Udishani NG, Bandara BR, Hara N, et al. Antifungal constituents of the stem bark of Bridelia retusa. Phytochemistry. 2003;62(4):637-641.
- Ghawate VB, Jadhav VS, Bhambar RS. Pharmacological activities of Bridelia retusa: a review. Pharmacology. 2015;9:415-418.
- Bhakuni DS, Dhar ML, Dhar MM, Dhawan BN, Gupta B, Srimal RC. Screening of Indian plants for biological activity: Part III. Indian J Exp Biol. 1971;9(1):91-102.
- Jadhav N, Kulkarni S, Mane A, Kulkarni R, Palshetker A, Singh K, et al. Antimicrobial activity of plant extracts against sexually transmitted pathogens. Nat Prod Res. 2015;29(16):1562-1566.
- Kumar T, Jain V. Antinociceptive and anti-inflammatory activities of Bridelia retusa methanolic fruit extract in experimental animals. Sci World J. 2014;2014:890151. doi: 10.1155/2014/890151
- Tatiya AU, Saluja AK, Kalaskar MG, Surana SJ, Patil PH. Evaluation of analgesic and anti-inflammatory activity of Bridelia retusa (Spreng) bark. J Tradit Complement Med. 2017;7(4):441-451.
- Itankar PR, Durugkar NG, Waikar SB, Saoji AN. Phytochemical Study of Bark of Bridelia retusa Spreng. Med Aromat Plant Abstr. 2003;25:229.
- Umar AK, Zothantluanga JH, Aswin K, Maulana S, Sulaiman Zubair M, Lalhlenmawia H, et al. Antiviral phytocompounds "ellagic acid" and "(+)-sesamin" of Bridelia retusa identified as potential inhibitors of SARS-CoV-2 3CL pro using extensive molecular docking, molecular dynamics simulation studies, binding free energy calculations, and bioactivity prediction. Struct Chem. 2022;33(5):1473-1490. doi: 10.1007/s11224-022-01959-3
- News Medical. Spike proteins [Internet]. 2022 [cited 2022 Jun 27]. Available from: https://www.news-medical.net/?tag=/Spike-Protein
- Shin D, Mukherjee R, Grewe D, Bojkova D, Baek K, Bhattacharya A, et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature. 2020;587(7835):657-662.
- Dassault Systèmes. Discovery Studio Visualizer [Internet]. 2022 [cited 2022 Jun 27]. Available from: https://discover.3ds.com/discovery-studio-visualizer-download
- PyRx. PyRx - Virtual Screening Tool [Internet]. 2022 [cited 2022 Jun 27]. Available from: https://pyrx.sourceforge.io/
- Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455-461.
- Dallakyan S, Olson AJ. Small-molecule library screening by docking with PyRx. Methods Mol Biol. 2015;1263:243-250.
- Umar A. Flavonoid compounds of buah merah (Pandanus conoideus Lamk) as a potent SARS-CoV-2 main protease inhibitor: in silico approach. Future J Pharm Sci. 2021;7(1):1-9.
- Arora S, Lohiya G, Moharir K, Shah S, Yende S. Identification of potential flavonoid inhibitors of the SARS-CoV-2 main protease 6YNQ: a molecular docking study. Digit Chin Med. 2020;3(4):239-248.
- CABS flex 2.0 [Internet]. Warsaw: Laboratory of Theory of Biopolymers; 2022 [cited 2022 Jun 28]. Available from: http://biocomp.chem.uw.edu.pl/CABSflex2/index
- Azam SS, Abbasi SW. Molecular docking studies for the identification of novel melatoninergic inhibitors for acetylserotonin-O-methyltransferase using different docking routines. Theor Biol Med Model. 2013;10(1):1-16.
- Zothantluanga JH. Molecular Docking Simulation Studies, Toxicity Study, Bioactivity Prediction, and Structure-Activity Relationship Reveals Rutin as a Potential Inhibitor of SARS-CoV-2 3CL Pro. J Sci Res. 2021;65(2):96-104.
- Zothantluanga JH, Gogoi N, Shakya A, Chetia D, Lalthanzara H. Computational Guided Identification of Potential Leads from Acacia Pennata (L.) Willd. as Inhibitors for Cellular Entry and Viral Replication of SARS-CoV-2. Future J Pharm Sci. 2021;7:201.
- Zothantluanga J, Aswin SK, Rudrapal M, Chetia D. Antimalarial Flavonoid-Glycoside from Acacia Pennata with Inhibitory Potential Against PfDHFR-TS: An In-Silico Study. Biointerface Res Appl Chem. 2021;12(4):4871-4887.
- Khan J, Al Asoom LI, Khan M, Chakrabartty I, Dandoti S, Rudrapal M, et al. Evolution of RNA Viruses from SARS to SARS-CoV-2 and Diagnostic Techniques for COVID-19: A Review. Beni-Suef Univ J Basic Appl Sci. 2021;10:60.
- Pasala PK, Abbas Shaik R, Rudrapal M, Khan J, Alaidarous MA, Khairnar SJ, et al. Cerebroprotective Effect of Aloe Emodin: In Silico and in Vivo Studies. Saudi J Biol Sci. 2022;29(2):998-1005.
- Umar AK, Zothantluanga JH. Structure-Based Virtual Screening and Molecular Dynamics of Quercetin and Its Natural Derivatives as Potent Oxidative Stress Modulators in ROS-Induced Cancer. Indones J Pharm. 2021;32(1):60-72.
- Patowary L, Borthakur MS, Zothantluanga JH, Chetia D. Repurposing of FDA Approved Drugs Having Structural Similarity to Artemisinin against PfDHFR-TS through Molecular Docking and Molecular Dynamics Simulation Studies. Curr Trends Pharm Res. 2021;8(1):14-34.
- Umar AK, Kelutur FJ, Zothantluanga JH. Flavonoid Compounds of Buah Merah (Pandanus Conoideus Lamk) as a Potent Oxidative Stress Modulator in ROS-Induced Cancer: In Silico Approach. Maj Obat Tradis. 2021;26(3):221-230.
- Rudrapal M, Celik I, Khan J, Ansari MA, Alarousy RMII, Yadav R, et al. Identification of Bioactive Molecules from Triphala (Ayurvedic Herbal Formulation) as Potential Inhibitors of SARS-CoV-2 Main Protease (Mpro) through Computational Investigations. J King Saud Univ Sci. 2022;34(3):101826.
- Rudrapal M, Celik I, Chinnam S, Ansari MA, Khan J, Alghamdi S, et al. Phytocompounds as Potential Inhibitors of SARS-CoV-2 Mpro and PLpro through Computational Studies. Saudi J Biol Sci. 2022;29(5):3456-3465. doi: 10.1016/j.sjbs.2022.02.028
- Zothantluanga JH, Abdalla M, Rudrapal M, Tian Q, Chetia D, Li J. Computational Investigations for Identification of Bioactive Molecules from Baccaurea Ramiflora and Bergenia Ciliata as Inhibitors of SARS-CoV-2 Mpro. Polycycl Aromat Compd. 2022;1-29.
- Pasala PK, Uppara RK, Rudrapal M, Zothantluanga JH, Umar AK. Silybin Phytosome Attenuates Cerebral Ischemia-reperfusion Injury in Rats by Suppressing Oxidative Stress and Reducing Inflammatory Response: In Vivo and in Silico Approaches. J Biochem Mol Toxicol. 2022;36(7):e23073. doi: 10.1002/jbt.23073
- Paul S, Hmar EBL, Zothantluanga JH, Sharma HK. Essential Oils: A Review on Their Salient Biological Activities and Major Delivery Strategies. Sci Vis. 2020;20(2):54-71.
- Zothantluanga JH. Ethnopharmacology and Phytochemistry-Based Review on the Antimalarial Potential of Acacia Pennata (L.) Willd. Sci Vis. 2020;20(4):139-147.
- Zothantluanga JH, Vanlalhriatpuii C, Lalthanzara H, Lalhlenmawia H, Bhat HR, Shakya A. Ethnomedicinal Plants Used Against Diarrhea Available in Mizoram, Northeast India: A Systematic Review. Sci Technol J. 2020;8(1):5-23.
- Zothantluanga JH, Lalnunpuii HS, Bhat HR, Shakya A. Awareness on the Possible Adverse Effects of Garcinia Cambogia: A Scientific Approach. Sci Vis. 2020;19(4):120-133.
- Zothantluanga JH, Bhat HR, Shakya A. A Systematic Review on the Nutraceutical Potential of Acacia Pennata (L.) Willd. Curr Trends Pharm Res. 2019;6(1):12-27.
- Pachuau L, Laldinchhana, Roy PK, Zothantluanga JH, Ray S, Das S. Encapsulation of Bioactive Compound and Its Therapeutic Potential. In: Advanced Pharmacological Uses of Medicinal Plants and Natural Products. IGI Global; 2021. p. 687-714.
- Sriwidodo, Umar AK, Wathoni N, Zothantluanga JH, Das S, Luckanagul JA. Liposome-Polymer Complex for Drug Delivery System and Vaccine Stabilization. Heliyon. 2022;8(3):e08934.
- Ghanbarzadeh S, Ghasemi S, Shayanfar A, Ebrahimi-Najafabadi H. 2D-QSAR Study of Some 2, 5-Diaminobenzophenone Farnesyltransferase Inhibitors by Different Chemometric Methods. EXCLI J. 2015;14:484-495.
- de Souza AS, Ferreira LLG, de Oliveira AS, Andricopulo AD. Quantitative Structure-Activity Relationships for Structurally Diverse Chemotypes Having Anti-Trypanosoma Cruzi Activity. Int J Mol Sci. 2019;20(11):2801.