
RESEARCH ARTICLE
Pharmacophore Modeling and Molecular Docking of Flavonoid Derivatives in Abelmoschus manihot Against Human Estrogen Receptor Alpha of Breast Cancer
Sciences of Pharmacy|Vol. 1, Issue 2, pp. 41-47 (2022)
CC BY 4.0-2022 Authors
Views
Downloads
Shares
Received
Aug 30, 2022Revised
Sep 21, 2022Accepted
Sep 26, 2022Published
Oct 3, 2022
Abstract
Tamoxifen is the most commonly used anti-estrogen adjuvant therapy for estrogen receptor-positive breast cancer. However, it is associated with an increased risk of some serious side effects, such as uterine cancer, stroke, and pulmonary embolism. The flavonoid compounds in the leaves of A. manihot inhibited the growth of 4T1 breast cancer cells at a CTC50 concentration of 185.06 μg/ml. Therefore, this study aims to examine the molecular interactions and pharmacophore modeling based on the interaction of 4-OHT with human ER, followed by the molecular docking of the flavonoid derivatives with human ERα. The molecular docking simulations and 3D structure-based pharmacophore models were used to identify the molecular interactions of flavonoid derivatives in A. manihot on estrogen receptors (ERα) (PDB ID: 3ERT). The results showed that the binding energies of the flavonoid derivatives in isorhamnetin and isoquercitrin were -8.68 kcal/mol and -8.75 kcal/mol, respectively. This compound also interacted with Arg394 and Glu353 important amino acid residues in the ERα-binding pocket. Meanwhile, the pharmacophore fit scores of isorhamnetin and isoquercitrin were 82.36% and 84.91%, respectively. The flavonoid derivatives in A. manihot had pharmacophore fit resulting from the 4-OHT complex with ER, and therefore they had potential as ERα antagonists. Out of the 10 flavonoid derivatives, isorhamnetin and isoquercitrin showed the best docking scores and could be used as candidates for new anti-breast cancer drugs with antagonistic activity against ERα.
Introduction
Cancer is currently the leading cause of death worldwide. In 2020, almost 10 million deaths were registered as a result of cancer, and according to report, breast cancer ranks first with 2.26 million cases and 685, 000 deaths (1). Estrogen receptors are the human prognostic marker used to identify tumors in breast tissue (2), and they consist of two subtypes, ERα and ERβ, which have different affinities for estrogen. ERα is a transcription regulator-activated ligand as a major regulator of breast differentiation and proliferation (3). Furthermore, it plays an important role in the development and progression of hormone-dependent breast cancer (4). Tamoxifen, as an antiestrogen can block estrogen signaling through a mechanism of competition with endogenous estrogens to bind to the estrogen receptor and alter its activity as a regulator of gene transcription. Additionally, it has antagonistic activity in the breast but is an agonist in the uterus and bone (5). Tamoxifen and its active metabolite 4-hydroxytamoxifen (4-OHT) have cytotoxic activity against MCF-7 breast cancer cells with an IC50 of 5 μM and 1 μM (6). However, the effectiveness of tamoxifen is limited by the presence of intrinsic resistance. Amplification and overexpression of COPS5 (complex subunit COP9) was a major cause of tamoxifen resistance in 86.7% of patients with ERα-positive breast cancer. Overexpression of COPS5 through isopeptidase activity leads to proteasome-mediated NCoR degradation, which is a major repressor of ERCC, and therefore, alternative medicine is needed (7).
Flavonoids are one of the promising natural remedies for breast cancer (8). One of the plants that contain active flavonoid compounds is Abelmoschus manihot L. Medik. Most people in Palu and Manado cities, Indonesia use the leaves as food, which were reported to have total flavonoids of 61.763% (9). It was reported that the flavonoid compounds in the leaves of A. manihot inhibited the growth of 4T1 breast cancer cells at a concentration of 50% inhibitory cell growth (CTC50) of 185.06 μg/mL (10). Furthermore, flavanoid compounds can act as selective estrogen receptor modulators (SERMs) as they are a group of compounds with a C6-C3-C6 chemical structure. SERMs can enter the cell and then bind to the ER, forming a complex bond. This complex binding binds to the Estrogen Response Element (ERE), which is located near the gene whose transcription is controlled. Additionally, the complex will activate nuclear receptor co-repressor (NCoR) protein and suppress cancer cell replication, therefore its proliferation can be controlled (11, 12). Currently, studies on pharmacophore and molecular docking of flavonoid derivatives in A. manihot to ERα are very limited. Therefore, it is necessary to conduct pharmacophore modeling tests based on the interaction of 4-OHT with human ERα followed by molecular docking of flavonoid derivatives of A. manihot with human ERα.
Experimental Section
Hardware and Software
The hardware used is a computer with Windows 10 64-bit OS, AMD A6-7310 APU Processor with AMD Radeon R4 Graphic 2.00 GHz and 6 GB RAM. The software is ChemDraw professional 15.0 (Academic License), LigandScout 4.4.5 Advanced (Universitas Padjadjaran License), AutoDock 4.2.6, AutoDockTools 1.5.6 (The Scripps Research Institute), and BIOVIA Discovery Studio 2021 (Academic License).
Preparation of Ligands and Molecular Docking
3D X-ray crystallographic structure of ERα in complex with 4-OHT (PDB ID: 3ERT) crystallized and stored by Shiau et al. , (1998) 1.90 Å resolution downloaded from the Protein Data Bank online (https://www. rcsb. org /structure/3ERT) (13). The ligands were separated from the receptor structure using the BIOVIA Discovery Studio 2021 R2 client. The 3D-mangostin structure and its derivatives as ligands were optimized by ChemOffice 2010 and ChemDraw Ultra 12.0 (PerkinElmer Inc. ) and LigandScout 4.4.5 Advanced (Inte: Ligand GmbH). Molecular docking simulations were carried out according to previous validation studies (14). ERα receptors and ligands were prepared for docking simulation using AutoDockTools 1.5.6. Furthermore, receptors as macromolecules are added with Kollman charges, while ligands are added with Gasteiger charges (15). The grid parameter file corresponds to a grid consisting of 40×40×40 points spaced 0.375Å and centered on the active edge ERα (x = 30, 010, y = -1, 913, and z = 24, 207). AutoDock 4.2.6 (The Scripps Research Institute) was used to perform molecular docking simulations. The docking parameter file corresponds to the Lamarckian Genetic Algorithm (LGA) with 100 run counts, 150 population sizes, 2, 500, 000 energy evaluations, 0.02 gene mutation rate, and 0.8 crossover rate (16). The conformational results from the simulation were grouped using the root mean square deviation (RMSD) tolerance of 1.0. The ligand conformation with the lowest free bond energy (∆G) was selected from the best cluster, and the best ligand conformation was used for the next analysis stage. The receptor-ligand complex from the docking simulation was visualized using the BIOVIA Discovery Studio Visualizer 2021. The determination of ligand interaction features for each pose in the receptor-binding pocket was analyzed with LigandScout 4.4.5 Advanced Inte: Ligand GmbH. Wina, Austria (8).
3D Structure-Based Pharmacophore Modeling
Structure-based 3D pharmacophore modeling was derived from an ERα X-ray structure complexed with 4-OHT (PDB ID: 3ERT) using Ligandscout 4.4.5 Advanced (17). Feature validation was performed by filtering 626 active sets and 20, 773 bait sets obtained from the Database of Useful Decoys (18). The flavonoid derivatives in A. manihot were screened virtually using a 3D structure-based pharmacophore model validated with the LigandScout 4.4.5 advanced algorithm. The result of this process is the adjustment value of the pharmacophore. The pharmacophore-fit score was used to measure the feature and geometry similarity of each achieved compound based on a 3D structure with a feature pharmacophore model with 4 feature counts removed for the combined pharmacophore, 10.0% optional threshold partial adaptation feature, and 1.0 feature tolerance scale factor.
Result and Discussion
Two hydrogen bonds were observed in the interaction between 4-OHT and human ERα. Hydrogen bonding was observed in the interaction between 4-OHT and human ERα bound to Glu353 as a hydrogen bond donor (HBD) and Arg394 as a hydrogen bond acceptor (HBA), as shown in Figure 1. The hydrophobicity of 4-OHT mostly interacts with the aromatic ring and the butenyl group also forms positive ionized interactions with the secondary amine nitrogen. Hydrogen bond interactions were formed as shown in Figure 2.
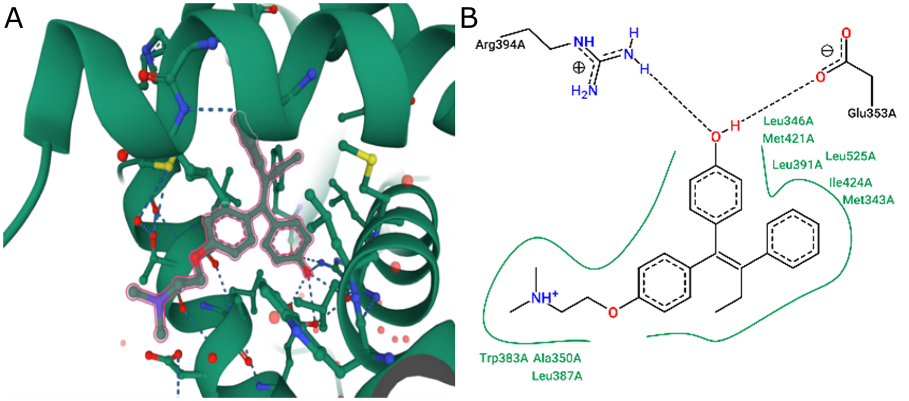
ERα has a ligand-binding domain (LBD) that is primarily a hydrophobic cavity composed of amino acid residues of helices 3, 6, 7, 8, 11, and 12. The agonist and antagonist activities of the ligands are determined by helix-12 of residue 536- 544 in its macromolecule (ERα). When a 4-OHT antagonist of ERα binds to LBD, helix-12 will be closed and not bound to the co-activator, and therefore it has antagonistic activity based on the absence of hydrogen bonding interactions with His524 (14). Based on the interaction between 4-OHT and the pharmacophore features of human ERα, two features of four aromatic rings of HBD and one HBA were produced using LigandScout.
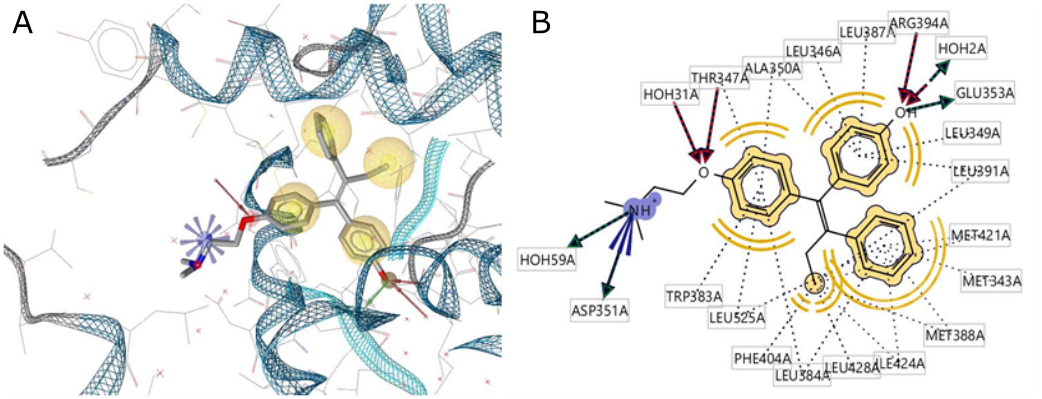
Molecular validation of the docking was performed by rebinding the co-crystallized 4-OHT to its original position at the human ERα binding site. The results show a binding mode similar to that of the original complex. The 4-OHT molecule interacts with Arg394 and Glu353. The validity of the docking program was confirmed by placing the 4-OHT pose, which was re-docking with the original resulting in an RMSD value of 0.590. This value is defined as valid as shown in Figure 3.
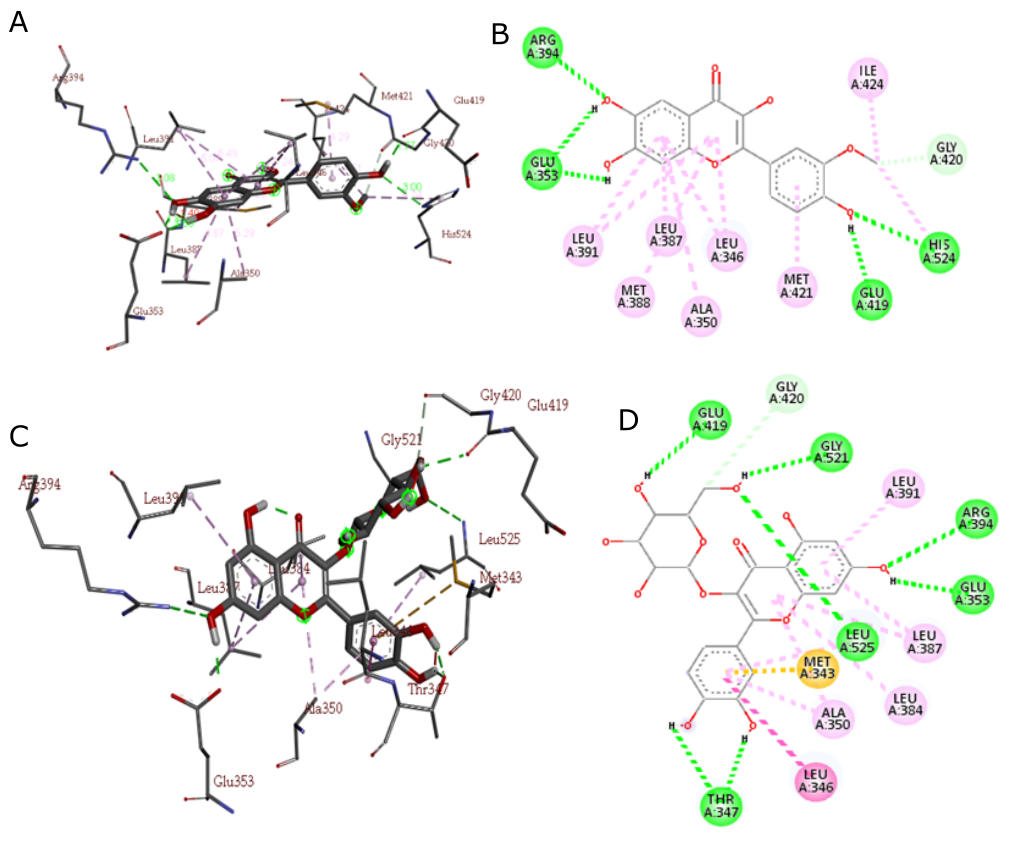
The best docking conformation of isorhamnetin and isokesitrin in the ERa ligand-binding domain is indicated by the presence of hydrogen bonds with Arg394 and Glu353 important amino acid residues in the human ERα binding pocket and with other amino acid residues. In addition to its interactions with Arg394 and Glu353, the binding conformation of isorhamnetin and isokesitrin within the ERα ligand-binding domain reveals significant hydrogen bonding with other key amino acid residues, as depicted in Figure 4. These interactions underscore the importance of these compounds in modulating the activity of ERα.
| No | MolecularNames | MolecularWeight | Log P | Number of Hydrogen Bond Donors | Number of Hydrogen Acceptors |
|---|---|---|---|---|---|
| 1 | Cannabiscitrin | 480.38 | 2.49 | 10 | 12 |
| 2 | Hibifolin | 494.37 | 4.58 | 9 | 14 |
| 3 | Hyperoside | 464.38 | 2.66 | 9 | 11 |
| 4 | Isoquecitrin | 464.38 | 2.66 | 9 | 11 |
| 5 | Quercetin | 302.24 | 4.22 | 6 | 6 |
| 6 | Isorhamnetin | 316.27 | 4.16 | 5 | 6 |
| 7 | Myricetin | 318.24 | 4.05 | 7 | 7 |
| 8 | Quercimeritrin | 464.38 | 2.66 | 9 | 11 |
| 9 | Quercitrin | 448.38 | 3.45 | 8 | 10 |
| 10 | Rutin | 610.53 | 2.23 | 11 | 15 |
The docking results in Figure 4 show that the dimethylaminoethoxy group of 4-OHT is more elongated than the methoxy and hydroxy groups of isorhamnetin and isoquecitrin. This difference can be explained by the lower binding free energy (∆G) of 4-OHT (−11.93 kcal/mol; inhibitory constant 1.79 nM) compared to G isorhamnetin (−8.68 kcal/mol; inhibitory constant 0.43 µM) and isoquecitrin (-8.75 kcal/mol; inhibitory constant 0.38 µM) (Table 2). The structural design of the flavonoid derivatives focused on the modifications of the methoxy group and the dihydroxy-substituted aromatic ring and was based on the key interactions between 4-OHT and ERα, according to Lipinski's Five rules as shown in Table 1.
| No | Molecule Name | G (Kkal/mol) | Ki (µM) | HB | HB Interaction with Arg394 or Glu353 |
|---|---|---|---|---|---|
| 1 | Cannabiscitrin | -7.37 | 3.96 | 5 | 2 |
| 2 | Hibifolin | -6.25 | 26.28 | 2 | 0 |
| 3 | Hyperoside | -8.28 | 0.85 | 4 | 2 |
| 4 | Isoquecitrin | -8.75 | 0.38 | 7 | 3 |
| 5 | Quercetin | -7.81 | 1.89 | 4 | 2 |
| 6 | Isorhamnetin | -8.68 | 0.43 | 5 | 3 |
| 7 | Myricetin | -7.34 | 4.18 | 3 | 1 |
| 8 | Quercimeritrin | -5.62 | 75.77 | 7 | 1 |
| 9 | Quercitrin | -8.54 | 0.54 | 3 | 1 |
| 10 | Rutin | -4.63 | 402.49 | 3 | 1 |
Results in Table 2 shows Lipinski's rule of five for orally administered drugs and this rule specifies four physicochemical parameters, namely molecular weight ≤ 500, log P ≤ 5, donor hydrogen bond ≤ 5, and acceptor hydrogen bond ≤ 10. These physicochemical parameters are related to the solubility and acceptable intestinal tract permeability and are part of the initial stage to determine the bioavailability of orally administered drugs (19).
The protein structure of hERα has hydrogen bonds in its constituent amino acid residues, namely between Glu419 and His524 and Glu419 with Lys531 (hydrogen bonding network). A disruption of this hydrogen-bonding network can be suggested by fluctuations in ligands that are potential antagonists of the hERα receptor (14). The ligands isorhamentin and isoquercitrin disrupt hydrogen bonds with fluctuations with residues Ser432 and Ser521, therefore it can be explained that isorhamentin and isoquercitrin may have antagonistic activity against hERα.
The validation of the pharmacophore model and the interaction properties based on 3D structures was performed by filtering 626 active sets and 20, 773 bait sets obtained from the Database of Useful Decoys (DUDe) (18). The results showed that the 100% enrichment factor (EF100%) was 32.4 with a 100% AUC of 1.00 as shown in the Receiver Operating Characteristic (ROC) curve (Figure 5). These results indicate that the 3D pharmacophore model can distinguish the active molecule from the feed molecule.
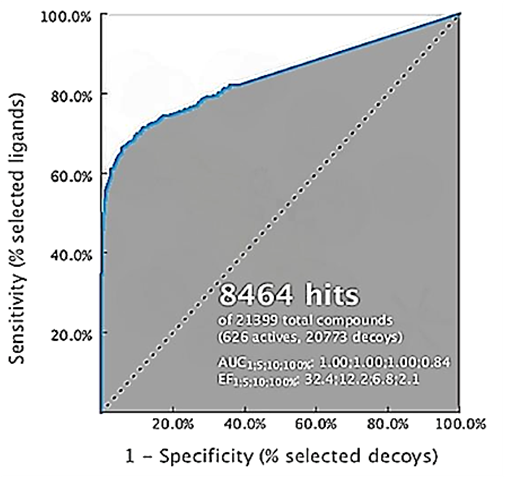
The pharmacophore fit score of the flavonoid derivatives in A. manihot is presented in Table 3. The pharmacophore fit score is a measure of the geometric similarity of the molecular features to the 3D pharmacophore model. The results indicate that isorhamnetin and isoquercitrin have high pharmacophore compatibility (82.36 and 84.91, respectively), meaning that the chemical properties of isorhamnetin and isoquercitrin are geometrically aligned with the chemical properties of 4-OHT.
Pharmacophore modeling can be used to determine the suitability score of pharmacophore against flavonoid derivatives in A. manihot. The pharmacophore fit score is the % measure of the geometric similarity of chemical features compared to the active 3D model of the pharmacophore ligand, namely tamoxifen (14). The results show that isorhamnetin and isoquercitrin have high pharmacophore compatibility values (≥50%) of 82.36% and 84.91%, respectively, which means that there are similarities in the chemical properties of isorhamnetin and isoquercitrin with the chemical properties of 4-OHT. Therefore, it was stated that isorhamnetin and isoquercitrin have a good affinity for hERα.
| No | MolecularNames | Docking Score (kcal/mol) | PharmacophoreFit-score |
|---|---|---|---|
| 1 | Cannabiscitrin | -7.37 | 83.12 |
| 2 | Hibifolin | -6.25 | 84.44 |
| 3 | Hyperoside | -8.28 | 84.75 |
| 4 | Isoquecitrin | -8.75 | 84.91 |
| 5 | Quercetin | -7.81 | 84.16 |
| 6 | Isorhamnetin | -8.68 | 82.36 |
| 7 | Myricetin | -7.34 | 84.84 |
| 8 | Quercimeritrin | -5.62 | 83.13 |
| 9 | Quercitrin | -8.54 | 84.86 |
| 10 | Rutin | -4.63 | 83.87 |

Conclusion
The flavonoid derivatives of A. manihot have similar pharmacophore properties resulting from the complexation of 4-OHT with ER, and therefore they have potential as ERα antagonists. Out of the 10 flavonoid derivatives in A. manihot, isorhamnetin and isoquercitrin showed the best docking scores and can be used as candidates for new anti-breast cancer agents with antagonistic activity against hERα.
Declarations
Acknowledgment
The authors thank the Pelita Mas Institute for the research grant and supports.
Conflict of Interest
The authors declare no conflicting interest.
Data Availability
The unpublished data is available upon request to the corresponding author.
Ethics Statement
Not applicable.
Funding Information
This study was funded by the Pelita Mas Institute with grant number of 018/YPM-STIFA-PL/LPPM/VI/2021.
References
- World Health Organization. https://www.who.int/news-room/fact-sheets/detail/cancer. 2020. (accessed, January, 2022)
- Sun YS, Zhao Z, Yang ZN, Xu F, Lu HJ, Zhu ZY, Shi W, Jiang J, Yao PP, Zhu HP. Review: Risk Factors and Preventions of Breast Cancer. International Journal of Biological Sciences. 2017;13(11):1387-1397. https://doi.org/10.7150/ijbs.21635
- Abdel-Hafiz HA. Epigenetic Mechanisms of Tamoxifen Resistance in Luminal Breast Cancer. Diseases. 2017;5(3):16. https://doi.org/10.3390/diseases5030016
- Bhatt S, Stender J, Joshi S. et al. OCT-4: a novel estrogen receptor-α collaborator that promotes tamoxifen resistance in breast cancer cells. Oncogene. 2016; 35: 5722–5734. https://doi.org/10.1038/onc.2016.105
- Day CM, Hickey SM, Song Y, Plush SE, Garg S. Novel Tamoxifen Nanoformulations for Improving Breast Cancer Treatment: Old Wine in New Bottles. Molecules. 2020; 25(5):1182. https://doi.org/10.3390/molecules25051182
- Gonzalez-Malerva L, Park J, Zou L, Hu Y, Moradpour Z, Pearlberg J, Sawyer J, Stevens H, Harlow E, LaBaer J. High-throughput ectopic expression screen for tamoxifen resistance identifies an atypical kinase that blocks autophagy. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(5):2058-2063. https://doi.org/10.1073/pnas.1018157108
- Lu R, Hu X, Zhou J, Sun J, Zhu A, Xu X, Zheng H, Gao X, Wang X, Jin H, Zhu P, Guo L. COPS5 amplification and overexpression confers tamoxifen-resistance in ERα-positive breast cancer by degradation of NcoR. Nature Communications. 2016; 7: 12044. https://doi.org/10.1038/ncomms12044
- Muchtaridi M, Syahidah HN, Subarnas A, Yusuf M, Bryant SD, Langer T. Molecular Docking and 3D-Pharmacophore Modeling to Study the Interactions of Chalcone Derivatives with Estrogen Receptor Alpha. Pharmaceuticals. 2017;10(4):81. https://doi.org/10.3390/ph10040081
- Sudewi S, Lolo WA, Warongan M, Rifai Y, Rante H. The ability of Abelmoschus manihot L. leaf extract in scavenging of free radical DPPH and total flavonoid determination. IOP Conference Series Materials Science and Engineering. 2017;259 1. https://doi.org/10.1088/1757-899X/259/1/012020
- Viani A, Wirawan A. Total Antioxidant and In-Vitro Cytotoxic of Abelmoschus Manihot (L.) Medik from palu of Central Sulawesi and Doxorubicin on 4T1 cells line and Vero Cells. Research Journal of Pharmacy and Technology. 2019;12(11): 5472-5476. https://doi.org/10.5958/0974-360X.2019.00949.1
- Girault I, Bieche I, Lidereau R. Role of estrogen receptor α transcriptional coregulators in tamoxifen resistance in breast cancer. Maturitas. 2006;54(4):342-351. https://doi.org/10.1016/j.maturitas.2006.06.003
- Liu, James H. Selective estrogen receptor modulators (SERMS): keys to understanding their function. Menopause: The Journal of The North American Menopause Society. 2020;27(10). https://doi.org/10.1097/GME.0000000000001585
- Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, Greene GL. The Structural Basis of Estrogen Receptor/Coactivator Recognition and the Antagonism of This Interaction by Tamoxifen. Cell. 1998;5(7):927-937. https://doi.org/10.1016/s0092-8674(00)81717-1
- Muchtaridi M, Yusuf M, Diantini A, Choi SB, Al-Najjar BO, Manurung JV, Subarnas A, Achmad TH, Wardhani SR, Wahab HA. Potential Activity of Fevicordin-A from Phaleria macrocarpa (Scheff) Boerl. Seeds as Estrogen Receptor Antagonist Based on Cytotoxicity and Molecular Modelling Studies. International Journal of Molecular Sciences. 2014;15(5):7225-7249.https://doi.org/10.3390/ijms15057225
- Alkandahri MY, Patala R, Berbudi A, Subarnas A. Antimalarial activity of curcumin and kaempferol using structure-based drug design method. Journal of Advanced Pharmacy Education and Research. 2021: 11(4): 86-90. https://doi.org/10.51847/q7yYE310JY
- Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of Computational Chemistry. 2009;30(16):2785-2791. https://doi.org/10.1002/jcc.21256
- Mysinger MM, Carchia M, Irwin JJ, Shoichet BK. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. Journal of Medicinal Chemistry. 2012;55(14):6582–6594.https://doi.org/10.1021/jm300687e
- Jorgensen WL, Duffy EM Prediction of drug solubility from structure. Advanced Drug Delivery Reviews. 2002;54(3):355-366. https://doi.org/10.1016/s0169-409x(02)00008-x
- Ramachandran B, Kesavan S, Rajkumar T. Molecular modelling and docking of small molecule inhibitors against NEK2. Bioinformation, 2016;12(2):62-68. https://doi.org/10.6026/97320630012062