
REVIEW
In Silico Studies of Drug Discovery and Design Against COVID-19 Focusing on ACE2 and Spike Protein Virus Receptors: A Systematic Review
Academic Editor: Ernest Domanaanmwi Ganaa
Sciences of Pharmacy|Vol. 2, Issue 3, pp. 171-183 (2023)
CC BY 4.0-2023 Authors
Views
Downloads
Shares
Received
May 11, 2023Revised
Jun 7, 2023Accepted
Jun 13, 2023Published
Jul 6, 2023
Abstract
The emergence of COVID-19 has prompted researchers worldwide to focus on developing drugs that specifically target ACE2 receptors and SARS-CoV-2 Spike Protein receptors. They have embraced an in-silico approach that employs virtual screening, molecular docking, and molecular dynamics to achieve this. This innovative method harnesses existing chemical and natural product databases to identify the most suitable ACE2 receptor blockers and SARS-CoV-2 Spike Protein inhibitors. By following the PRISMA statement guidelines, a thorough literature search yielded 21 relevant articles, forming the basis of this systematic review. The review provides a comprehensive summary and detailed description of the methodologies, protocols, software tools, and noteworthy drug candidates identified in these studies. Additionally, it sheds light on the crucial molecular interactions by presenting an overview of the interacting residues elucidated in the reviewed articles, offering valuable insights for effective therapeutic interventions. Furthermore, the review presents thought-provoking suggestions for future research directions, aiming to inspire and guide advancements in drug development efforts.
Introduction
Coronaviruses are enveloped RNA viruses that are widely distributed among humans and other animals and can cause respiratory, enteric, and hepatic diseases in most cases (1). Severe Acute Respiratory Syndrome – Corona Virus (SARS-CoV) was the primary cause of severe acute respiratory syndrome outbreaks in Guangdong Province, China, in 2002 and 2003. In 2004, several reports from laboratory and nonlaboratory cases highlighted the possibility of SARS re-emergence (2). In late December 2019, there was a report about emerging pathogens linked to coronaviruses. Local health facilities in Wuhan reported several clusters of patients with pneumonia of unknown origin linked to a seafood and wet animal wholesale market in Hubei Province, China (3). Previous research in viral diagnostics, isolation and identification has revealed that the novel virus shares more than 85% identity with a bat SARS-like CoV genome. Further analysis of the 2019-nCoV genome from clinical specimens reveals 86.9% nucleotide sequence identity to a previously published bat SARS-like CoV. Although it is similar to some beta coronaviruses found in bats, it is not the same as SARS-CoV (4). This study served as the foundation for many subsequent studies on novel coronavirus drug discovery and vaccine development.
Another critical research is on the matter of viral receptors in the human body. SARS-CoV-2 uses human Angiotensin-converting Enzyme 2 (ACE2) as an entry receptor with similar affinity to previous SARS-CoV isolates, according to Walls et al. (5), indicating that the virus could spread effectively in humans. Another study by Zhou et al. (6) found that SARS-CoV-2 can use all ACE2 proteins, except mouse ACE2, as an entry point to enter cells that express ACE2, but not those that do not, indicating that ACE2 is the cell receptor through which the virus can enter the cells. They also show that SARS-CoV-2 does not use previous SARS-CoV receptors such as aminopeptidase N (APN) and dipeptidyl peptidase 4 (DPP4).
Drug discovery and development is a complicated process that needs extensive research and also has high-risk factors of failure (7). Conventional drug discovery methods could take more than a decade to complete, and the cost of research could be prohibitively expensive, which could reach around $70 mil- lion for each NME reaching the clinic (8). The failure that could occur in the process namely lack of clinical efficacy, unmanageable toxicity, poor drug-like properties and lack of commercial needs (9). Computational methods such as virtual screening and molecular docking could assist in the early stages of discovery (10). Modern computational-aided drug design is divided into two parts based on the molecular information source utilized, which are structure-based drug design and ligand-based drug design (11). Compounds from the existing database or new chemical entities could be screened more quickly to find the hit and lead compound, which could then be developed further to become the final drug (12).
In this article, the authors conducted a systematic review of drug discovery research and methods by using computational methods to find drug candidates from drug repurposing or a database of chemical entities. The authors systematically selected documentation on this subject and compared different computational analysis methods towards the ACE2 receptor or Spike Protein SARS-CoV-2 receptor in the current study.
Materials and Methods
Study Protocols
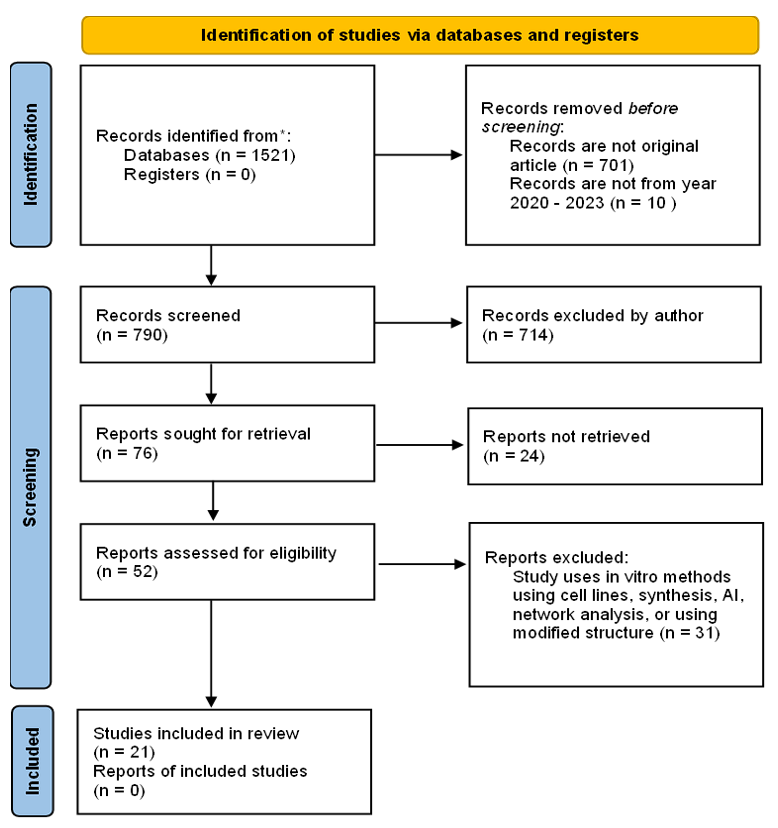
The Preferred Reporting Items for Systematical Reviews and Meta-Analysis (PRISMA) statement is used as a guideline to conduct the literature search (13)”, “computational”, “virtual screening”, and “molecular docking” using the Boolean operator. The search was conducted in ScienceDirect. The potential studies were screened according to the inclusion and exclusion criteria. The study flow can be seen in Figure 1.
Inclusion and Exclusion Criteria
The inclusion criteria for this article review were: (1) experimental studies written in English, (2) studies that use virtual screening or molecular docking as their drug discovery method, (3) databased used in the studies are existing drug databases or databases of natural product with non-peptide small molecule and (4) studies are published in the range year 2020 - 2023.
The exclusion criteria were: (1) studies written in other languages besides English, (2) non-related studies such as: (a) in vitro studies using cell line, (b) synthesis method, (c) computational using artificial intelligence, (d) network analysis method, (e) screening or docking using modified structure, (3) duplicate publications, (4) articles not available as full text.
Selection of Studies and Data Collection
All search results were collected and screened. All identified studies are assessed based on the title and abstract without using any specific data extraction form. The data are gathered according to the year of publication, computational method, and database used in the study.
Quality of Article Assessment Study
The idea of bias in computational drug research studies is not well established, therefore it is hard to determine the risk of bias in this type of study. The authors decided to go with a tool designed for the assessment in a similar in silico review published by Mohamed et al. (14) with some refinement. The assessment encompassed five main aspects of the quality of studies included in the present systematic review: design (single-target or bi-target or multi-target), target template and crystal resolution, docking tools, molecular dynamics simulation (yes or no) and the resource of the database. The quality of each eligible article was appraised by the author.
Results and Discussion
Literature Collection Process
The keywords used in the literature search yielded 790 research articles published between 2020 and 2023. Only 76 of those articles met the criteria for related studies. We retrieved 52 potential literature articles from the 76 articles and excluded 24 articles because they were written in a language other than English, the method used was unclear, or it was a review article that was missed in the first screening. We evaluated the retrieved articles for eligibility and decided to exclude 31 records that used a modified structure for docking, an in vitro method, a synthesis method, AI simulation, and network analysis. As a result, 21 articles were identified as meeting the inclusion criteria and were given full consideration. The final 21 articles are processed to the Quality of Article Assessment Study.
Quality of Article Assessment Study and Summary of Studies
We have summarized the quality of articles in our studies in Table 1. Bias in computational drug research studies is not well established, as stated in the methods. But one potential bias is the target's molecular plasticity.
The target's flexibility is usually limited or ignored when only molecular docking is used in a study. In this case, MD simulation could play a key role in drug discovery and design, as well as pre-and post-docking simulation. It can be used as a generator for multiple target conformations for virtual screening, or as a validator for post-docking to distinguish between improper docking poses and meaningful ones (15). 13 of our reviewed papers have been validating further by using various molecular dynamics programmes such as GROMACS (16), UNRES (17), NAMD (18), Schrodinger Desmond (19), and AMBER (20).
The majority of the reviewed articles use GROMACS and Schrodinger Desmond as their methods of validation and use various configurations for the simulation. 9 articles used a single target as their receptors, which is either ACE2 or the SARS-CoV-2 Spike Protein (SP), while 6 of the articles use a single target but with addition other than ACE2 or the SARS-CoV-2 SP. One article used exactly double targets which are ACE2 and SARS-CoV-2 SP, and the last 5 use both targets with the addition of another target.
These other targets are proteins which also contribute to the infection or multiplication process of the virus such as Main protease (Mpro)/3-chymotrypsin-like protease (3CLpro), papain-like protease (PLpro), RNA-dependent RNA polymerase (RdRp), and other non-structural protein (Nsp) for example Nsp-3 and Nsp-9. Crystal resolution is also one of the crucial parameters in molecular docking and dynamics studies. In this review, all the crystal structures used in the studies range from 2 Å to 4 Å, considered adequate.
Resolution is defined as a metric for assessing the quality of data collected on the protein or nucleic acid crystal. It evaluates the level of detail in the diffraction pattern as well as the level of detail that will be visible when the electron density map is computed (42). Higher resolution values, such as 1Å, are highly ordered and allow you to see every atom in the electron density map, whereas lower resolution values, such as 3Å or higher, only show the basic contours of the protein chain (43, 44).
The docking tools mainly used in the reviewed literature are Autodock4 (45) and Autodock Vina (46), though other tools are being used such as GLIDE (47), Cresset Flare (48), or MOE (49). The structure of the database mainly comes from PubChem (50), but other databases such as DrugBank (51), Ambinter (52), and ChemDiv (53) are also being used.
| No. | First Author, Year | Targets | Target Template | Crystal Resolution (Å) | Docking Tools | MD Simulation | Resource of structureDatabase | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sgl | Sgl+ | Dbl | Dbl+ | Spike Protein | ACE2 | Spike Protein | ACE2 | Yes | No | ||||
| 1 | Gurung, 2022 (21) | 7WBL | 3.40 | Autodock 4.2 | Gromacs | PubChem | |||||||
| 2 | Kulkarni, 2020 (22) | 6M0J | 2.45 | Autodock Vina | PubChem | ||||||||
| 3 | Rameshkumar, 2021 (23) | 6VW1 | 2.68 | Autodock Vina | Var. | ||||||||
| 4 | Natesh, 2021 (24) | 6W41 | 1R42 | 3.08 | 2.20 | Autodock Vina | PubChem | ||||||
| 5 | Al-shuhaib, 2022 (25) | 1R42 | 2.20 | GLIDE | UNRES | PubChem | |||||||
| 6 | Hadni, 2022 (26) | 6M17 | 2.90 | Autodock 4.2 | NAMD | PubChem | |||||||
| 7 | Kar, 2022 (27) | 6M0J | 2.45 | Autodock Vina | Gromacs | PubChem | |||||||
| 8 | Vardhan, 2020 (28) | 2GHV | 6M17 | 2.20 | 2.90 | Autodock Vina | CABS-flex 2.0 | Var. | |||||
| 9 | Kiran, 2022 (29) | 6VSB | 3.46 | Cresset Flare Docking | PubChem | ||||||||
| 10 | Jain, 2021 (30) | 6M0J | 2.45 | Autodock Vina | Gromacs | PubChem | |||||||
| 11 | Khan, 2021 (31) | 6M0J | 2.45 | Autodock Vina | Gromacs | PubChem | |||||||
| 12 | Benítez-Cardoza, 2020 (32) | 1R42 | 2.20 | Autodock + MOE | EXPRESS-pick | ||||||||
| 13 | Gowrishankar, 2021 (33) | 6VYB | 1R42 | 3.20 | 2.20 | Autodock 4.2 | Gromacs | PubChem | |||||
| 14 | Muhseen, 2020 (34) | 6LZG | 2.50 | Autodock Vina | Gromacs | NPACT + MPD3 | |||||||
| 15 | Akinlalu, 2021 (35) | 6LZG | 2.20 | Autodock Vina | DrugBank | ||||||||
| 16 | Baby, 2021 (36) | 1R4L | 3.00 | GLIDE | Schrodinger Desmond | DrugBank | |||||||
| 17 | Pokhrel, 2021 (37) | 1R4L | 3.00 | Autodock 4.2 | Schrodinger Desmond | Ambinter | |||||||
| 18 | Yu, 2022 (38) | 7DF4 | 7DF4 | 3.80 | 3.80 | Autodock Vina | TCM | ||||||
| 19 | Singh, 2022 (39) | 6LZG | 6M0J | 2.50 | 2.45 | Autodock 4.2 | Gromacs | PubChem | |||||
| 20 | Mishra, 2021 (40) | 6M0J | 6M0J | 2.45 | 2.45 | GLIDE | Schrodinger Desmond | PubChem | |||||
| 21 | Nabati, 2022 (41) | 6M0J | 2.45 | Autodock Vina | ChemDiv | ||||||||
Table 2 provides a comprehensive summary of the included studies, highlighting the methods, protocols, software, ligand candidates, targets, grid sizes in virtual screening and molecular docking, ADMET/physicochemical analysis software, and key interacting residues. Moreover, the table also delineates the important residues implicated in the molecular interactions under investigation, providing valuable insights into the specific amino acids or functional groups involved in driving the observed effects. This information serves as a valuable resource for understanding the research parameters and findings. In subsequent subsections, we discuss each parameter in detail, offering insights into the methodologies employed and their implications. This comprehensive analysis aims to provide a deeper understanding of the reviewed studies, shedding light on the approaches utilized and their impact on the field of research.
Virtual Screening
There are millions of chemical 'libraries' that a trained chemist could hope to synthesise. Combinatorial chemists have already demonstrated in several prototype systems that libraries containing 1, 000-100, 000 compounds can be assembled (54). Virtual screening help chemist decides what compound should be synthesised (55). Virtual screening can be done by docking method or pharmacophore method.
Pharmacophores are the "refined" essence of what makes an effective ligand-receptor interaction, explicitly three-dimensional, and represent fundamental physicochemical aspects of ligand-receptor interactions, and are extremely useful when experimental structural data is unavailable and homology models are unreliable. In that case, a good pharmacophore model could give powerful insight and screen more effectively (56, 57). Since the conformer is only compared against a three-point pharmacophore model, the method of virtual screening using pharmacophore could be very useful for a large number of compounds when compared to virtual screening using the docking method (54). This is due to the memory used for each conformation is not as large relative to when docking is required.
This method of virtual screening using pharmacophore could be seen in the work of Pokhrel et al. (37) which screened more than 11 thousand from the Ambinter database. The complicated steps in the pharmacophore model method are in the step of model validation. The literature used the GH scoring method for pharmacophore model validation and also includes enrichment factor and goodness of hit score. This validation needs to use a set of other databases called decoy compounds, which are usually available in the Database of Useful Decoys (DUDe) (58). This database has to be compared to active compounds in this matter they use known active ACE2 inhibitors from CheMBL (59) and a literature search. After the model is validated, then it can be used as the parameter for the screening. This could be a problem when there is no available decoy database, or the number of active compounds is inadequate.
| Ref. | Method, Protocol | Software | Ligand | Target | Grid Size (Å) | MD | ADMET | Candidate Drugs (ΔG) (kcal/mol) | Std. Ref. | Important Residue |
|---|---|---|---|---|---|---|---|---|---|---|
| (21) | PISA, MolDocPhysPropMD | PDBsum, ADT 1.5.6, AD4.2, DW 4.6.1, GROMACS 2019.2, LigPlot+ v1.4.5 | 36 compounds with a preclinical or clinical trial against previous variants | SP Omicron - hACE2 (7WBL) | 36 x 52.875 x 57.75 | 100ns300K | DW 4.6.1 | Abemaciclib (-10.08), Dasatinib (-10.06), Spiperone (-9.54) | - | SP: Phe338, Asp339, Asp364 |
| (22) | Act. Site, MolDoc, DFT | PyRx, ADV | Major components of essential oils | ACE2-RBD (6M0J) | (adjusted according to active site residues that are selected) | - | - | Anethole (-5.2), Cinnamaldehyde (-5.0), Carvacrol (-5.2), Geraniol (-5.0), Cinnamyl acetate (-5.2), L-4-terpineol (-5.1), Thymol (-5.4), Pulegone (-5.4) | - | SP: Arg454, Ser459, Glu471, Tyr505 |
| (23) | VS, MolDoc, PhysProp DL | ADV, AD4.1, CoDockPP, SA | 458 flavonoid compounds | SP Omicron - hACE2 (6VW1), Mpro (6LU7), RdRp (6M71) | 40 x 40 x 40 | - | SA | Albireodelphin (-11.2), Amentoflavon (-10.2), Cupressuflavon (-10.0), Agathisflavone (-9.9) | - | SP: Thr319, Thr394, Phe396, Arg553, Lys621, Asn628, Asp760, Asp761 |
| (24) | MolDoc, ADMET | ADTADV, ADMETLab, PT2, OSIRIS Property | Standard drugs and spices | Spike Protein (6W41), ACE2 (1R42), Mpro (6LU7) | (Interacting critical residues in Spike Protein and ACE2 complex) | - | AL, PT2, OSIRIS | Bioactives in asafoetida and sesame seed | Remdesivir | ACE2: His34, Glu37, Asp38, Arg393SP: Arg403, Gln493, Ser494 |
| (25) | MolDoc, ADMET, MD | GLIDE, SA, UNRES online server | 3392 compounds from Iraqi medicinal plants | ACE2 (1R42) | 30 x 30 x 30 | 250ns300K | SA | Epicatechin (-6.05) | - | ACE2: Asp30, Asn33, His34, Glu37 |
| (26) | MolDoc, ADMET, MD | ADT, AD4.2, DSV. | Bioactive flavonoids compounds | Spike Protein RBD (6M17), 3CLpro (6LU7) | 60 x 60 x 60 | 100ns310K | ADMET parameters | Herbacetin (-8, 03), Morin (-8, 46), Silibinin (-9, 03), Tomentin E (-8.32), Amentoflavone (-10.19), Bilobetin (-8, 89), Baicalein (-8.19), Quercetin (-8.26) | - | SP: Glu35, Asp38, Lys353, Glu406, Try453, Ser494, Gly496, Asn501, Tyr505 |
| (27) | MolDoc, ADMET, MD, PCA | ADV, ADT, SApkCSM, LigPlot+. PLIP. GROMACS 2018.3 | 300 compounds from 25 Indian medicinal plants | ACE2-RBD (6M0J), Mpro (6LU7) | 50 x 50 x 50 | 100ns300K | SA, pkCSM | Oleanderolide (-8.3), Proceragenin A (-8.3), Balsaminone A (-8.3) | - | SP: Cys336, Gly339, Asn343, Ala348, Arg355. Ser373, Asp428, Thr430 Phe515, |
| (28) | MolDoc, MD, ADMET, PLI | ADV, pkCSM, Mi, CABS-flex 2.0 online, | 154 phytochemicals in Limonoids and Triterpenoids class | SP (2GHV), ACE2 (6M17), 3CLpro (6LU7), Plpro (4MM3), RdRp (6M71) | - | 10ns | pkCSM Mi | SP: Maslinic acid (-9.3), Glycyrrhizic acid (-9.3), Corosolic acid (-9.4)ACE2: Glycyrrhizic acid (-9.5), Maslinic acid (-8.5), Obacunone (-8.1) | - | ACE2: Arg273, His345, Arg393 |
| (29) | MolDoc, SAP, ADMET. | Cresset Flare Docking, LigPlus, pkCSM | 37 phytoconstituents from K. Kudineer Chooranam and JACOM | Spike Protein (6VSB) | (based on trial and error) | - | pkCSM | Chrysoeriol (-11.39), Luteolin (-11.15), Quercetin (-11.47) | - | SP: Cys336, Phe338, Gly339, Phe342, Asn343, Thr345, Asp364, Val367, |
| (30) | BSP, MolDoc, MD | CASTp, Rampage, PyRx 0.8, GROMACS 2020, LigPlot+, VMD | 10 dietary flavonoid compounds | Spike Protein (6M0J) | 44.34 x 70.98 x 44.58 | 1ns300K | - | Naringin (-9.8) | Dexa-methasone | *SP: Asp367, Thr371, Glu406, Ser409, Lys441 |
| (31) | MolDoc, MD | UCSF Chimera, Autodock Vina, GROMACS 5.1 | 24 drug molecules | ACE2 (6M0J) | - | 10ns300K | - | Cefpiramide (-9.1) | - | - |
| (32) | VS, MolDoc, FE | MOE, AD, PT2 | 500, 000+ small molecules from Chembridge Corp. | ACE2 (1R42) | - | - | PT2 | Chem7781334 (-5.87), Chem7676800 (-5.84), Chem7956590 (-5.83) | - | ACE2: Gln24, Asp30, His34, Tyr41, Gln42, Met82, Lys353, Arg357 |
| (33) | MolDoc, PK, MD | DSV, AD4.2, Mi, admetSAR, GROMACS 5.1, | 57 phyto-ligands from Indian Herbs | ACE2 (1R42), Spike Protein (6VYB) | ACE2: 22.5 x 22.5 x 22.5 SP: 30 x 28.125 x 25.5 | 25ns300K | Mi, admetSAR | ACE2: Apigenin-7-O-glucuronide (-8.8), Ellagic acid (-8.4), Vasicolinone (-7, 5), SP: Apigenin-7-O-glucuronide (-7.2), Ellagic acid (-6.2), Vasicolinone (-6, 4) | - | ACE2: Lys26, Gln89 SP: Tyr28, Tyr269, Asp290 |
| (34) | MolDoc, ADMET, MD, PCA, FE | ADTADV, SA, pkCSM, GROMACS 5.1 | 1000 plant bioactive terpenes compound | Spike Protein (6LZG) | 45 x 45 x 45 | 50ns300K | SA, pkCSM | NPACT01552 (-11.0), NPACT01557 (-10.3), NPACT00631 (-9.5) | - | Tyr449, Tyr453, Glu484, Gly496, Gln498, Asn501 |
| (35) | Act. Site, MolDoc, ADMET. | DSV, UCSF Chimera, ADTCASTp, PyRx, ADV, SA, AdmetSAR, GraphPad Prism | 791 FDA-approved drugs | ACE2 (6LZG), 3CLpro (6LU7), ADP ribose phosphatase of NSP3 (6VXS), NSP9 RNA binding protein (6W4B) | (residues on active site included) | - | SA, AdmetSAR | Ethynodiol diacetate (-15.6), Methylnaltrexone (-15.5), Ketazolam (-14.5), Naloxone (-13.6), | Lopinavir, Remdesivir, Hydroxy-chloroquine | Ile291 |
| (36) | MolDoc, MD | Protein Prep Wizard, Schrodinger's HTVS, Glide SP, Schrodinger Desmond platform | 2800 FDA-approved drugs | ACE2 (1R4L)TMPRSS2 (Hom. Mod. ) | (Glide grid tool) | 50ns300K | - | Valrubicin (-8.59), Lopinavir (-7.89), Fleroxacin (-7.73), Alvimopan (-8.51), Arbekacin (-7.74), Dequalinium (-7.73) | - | Arg273, Lys363, Asp367, Thr371, |
| (37) | VS PharmacophoreMolDoc, ADMET, MD, FE | LigandScout 4.3, DSV 16.1, PyRx, AD4, ADV, SA | 11, 295 compounds from the database | ACE2 (1R4L) | 20.05 x 17.92 x 8.75 | 250ns300K | SA | Amb17613565 (-7.5), Amb6600091 (-7.1), Amb3940754 (-7.1), Amb21855906 (-7.0) | XX5 | Asn149, Gly268, Asp269, Arg273, Asn277, Asp350, Lys363, Thr365, Asp367, Arg393 |
| (38) | MolDoc | ADV, M2M | 28 natural plants from TCM | ACE2 (7DF4), Spike Protein (7DF4) | (Interacting critical residues in Spike Protein and ACE2 complex) | - | - | ACE2: Oleanolic acid (-7.1), Tryptanthrin (-6.55), Chrysophanol (-6.16), Rhein (-4.69)SP: Oleanolic acid (-3.74), Tryptanthrin (-4.26), Chrysophanol (-4.07), Rhein (-3.66) | - | ACE2: His34, Lys94, Gln102, Lys562, Trp566 SP: Arg403, Tyr449, Tyr453, Gln493, Ser494, Tyr495, Gly496 |
| (39) | ADMET, MolDoc, Qua. Calc, MD, | SA, admetSAR, UCSF Chimera, AD4.2, DSVGaussian16 suite, GROMACS 2015, | 586 phytochemicals from 47 medicinal plants | ACE2 (6M0J), Spike Protein (6LZG), Plpro (6W9C), Mpro (6LU7), Importin α-5 (2JDQ), Importin β-1 (1F59) | 33.75 x 33.75 x 33.75 | 50ns300K | SA, AdmetSAR | ACE2: Hetisinone (-8.46) SP: 14-deoxy-11, 12-didehydroandrographolide (-7.76) | Arbidol (SP), Hydroxychloroquine (ACE2) | ACE2: Ala348, Trp349, His378, His401 SP: Arg23, Ser182, Phe183, Leu185 |
| (40) | MolDoc, FE, MD. | Schrodinger Drug Discovery Suite, Pymol 2.4.1 | 85 antiviral and antimicrobial flavonoid compounds | ACE2 (6M0J), Spike Protein (6M0J), Mpro (6LU7), NSP12 (7BV2), NSP15 (6WXC) | 30 x 30 x 30 | 100ns300K | - | SP: Isosilybin (-5.19)ACE2: Legalon (-2.78) | - | SP: Tyr453, Gly496, Gln498, Tyr505ACE2: Lys26, Glu37, Asn90, Gln96, Ala387, Arg393 |
| (41) | VS, MolDoc, ADMET | Chimera 1.13, Molegro Virtual Docker v6.0, PyRx 0.8, DSV, ADV, ADT1.5.6, OpenBabelSA, ADMETlab 2.0 | 100, 000+ chemical compounds from ChemDiv | Spike Protein (6M0J), Mpro (7AMJ), RdRp (7B3D), Plpro (6WX4) | 23.60 x 45.00 x 21.88 | - | SwissADME, ADMETlab 2.0 | 8008-2051 (-9.0)K279-0710 (-8.8) | Nebivolol | Arg403, Tyr449, Tyr453, Ser494, Gly496, Gln498, Asn501, |
Molecular Docking
The docking method is another option for virtual screening. It can also further be used to validate virtual screening results. Molecular docking studies are mainly used to predict the ligand-receptor complex's binding affinity, preferred binding pose, and interaction with the least amount of free energy. Docking studies also can reveal the interaction between protein-ligand, protein-nucleotide, and also protein-protein interactions (PPIs). Noncovalent interactions can include ionic bonds, hydrogen bonds, and van der Waals interactions (60). In addition to the software mentioned previously in the summary of studies, several other software options are widely used in many molecular docking studies. RosettaLigand (61), Surflex (62), and Ligandfit (63) are some of the other popular software.
The docking mechanism is a two-step mechanism. It started with sampling conformation of the ligand in the receptor then followed by ranking these conformations using a scoring function. The effectiveness of a docking programme is determined by two major factors: search algorithms and scoring functions (64). There are 2 main algorithms in molecular docking, which is the stochastic algorithm when where the search is carried out by modifying the ligand conformation or population of ligands. Example algorithms for this method are Monte Carlo and Genetic Algorithms (65). On the other hand, systematic search methods promote minor modifications in structural parameters, which gradually change the conformation of the ligands. The algorithm examines the energy landscape of the conformational space and, after many search and evaluation cycles, converges on the lowest energy solution corresponding to the most likely binding mode. The systematic algorithms are presented in GLIDE and DOCK (66).
Scoring functions are used to predict the target-ligand complex's binding free energy, which is a measure of the small molecule's binding potency for the biomolecular target. Scoring functions are classified into three types: force-field-based scoring, empirical-based scoring, and knowledge-based scoring (67). AutoDock scoring is an example of force-field-based scoring, which is derived from the classic force field and evaluates the binding energy as a sum of nonbonded interactions. Empirical-based scoring, such as GlideScore, is a weighted sum of various types of receptor-ligand interactions. DrugScore is a knowledge-based scoring system that penalises repulsive interactions while favouring preferred contact between each of the atoms in the protein and ligand within a given cut-off (64).
Grid Size and Parameter
In our reviewed studies, we examined several variations in the grid size parameter. Several studies reported the grid size qualitatively, stating that they used active site residues or critical interacting residues as grid size (22, 24, 35, 38). Because of the lack of quantitative data, this approach of disclosures would, of course, reduce the reproducibility of the studies. In other studies, the grid size is determined differently for each receptor target (33). In others, it is the same size for all tested receptors (23, 25-27, 39-41). Understandably, a specific PDB will have a set of specific grid sizes that can be used in it, but the grid size should always be bigger than the ligand that is docked. According to Feinstein and Brylinski's research, the highest accuracy is obtained when the dimensions of the search space are 2.9 times larger than the radius of the gyration of a docking compound (68). They developed a procedure based on this discovery to customise the box size for individual query ligands to maximise docking accuracy. This finding essentially reduces the number of scoring failures caused by overly generous box sizes while also avoiding sampling failures caused by a too-narrow search space.
Database of Chemicals
Although the majority of the chemical structure is obtained from PubChem, the dataset used for each study is unique to each author. Several studies are being conducted to investigate whether already approved drugs can be repurposed from their original purpose to become ACE2 blockers and SARS-CoV-2 inhibitors (31, 35, 36). Another method for preparing the dataset for testing is to look for compounds found in traditional medicine or plant constituents as drug candidates (22, 24, 39, 40, 25, 27-30, 33, 34, 38). The final notable approach is to select a chemical suitable as a candidate from a large dataset ranging from 10, 000 to 500, 000 compounds (32, 37, 41). However, these differences in approach may provide beneficial insight from different perspectives, bringing us closer to real drug candidates ready for development.
Target Receptors
We may find a range of PDB IDs for ACE2 and SARS-CoV-2 Spike Protein receptors in this review study. The same PDB ID was used in several articles for the same receptors. We can see this in four publications (24, 25, 32, 33) for ACE2, which used PDB 1R42 (69) as the receptors. Some other interesting point is that a PDB file can be used as a receptor target for ACE2 or SARS-CoV-2 because it contains both receptors in one file. We can see the PDB 6M0J (70) can be used as a SARS-CoV-2 Spike Protein target in 5 articles (22, 27, 30, 40, 41) and for ACE2 in 3 other articles (31, 39, 40). Both mentioned receptors are good in crystal resolution (2.20 and 2.45 respectively).
Interacting Residues
We summarised the common residues that appear more than once in the reviewed literature from the reported residues that interact with the ligands. Tyr453 and Gly496 appear in four different articles in the SARS-CoV-2 Spike Protein, while Arg403, Ser494, and Tyr505 appear in three distinct articles. Several residues appear in two articles: Cys336, Phe338, Gly339, Asn343, Asp364, Ser373, Tyr449, Gln493, and Asn501. The binding residues in the ACE2 receptor that appear more than once in the reviewed article are as follows. His34 was mentioned in five different articles. Glu37 and Arg393 were mentioned in four different articles. Three different articles featured Asp30, Arg273, and Asp367. While Lys26, Asn33, Lys363, and Thr371 were found in two separate articles. These residue similarities could help researchers find more specific binding sites, as well as serve as a reference for future drug discovery and development aimed at ACE2 and SARS-CoV-2.
Molecular Dynamics
Advances in software and hardware performances allowed researchers to adopt molecular dynamics to address drug discovery issues, especially protein–ligand stability (71). The dynamic nature of the receptor has been largely demonstrated and conformational changes have been related to ligand binding (71). In brief, MD simulation begins with the selection and preliminary analysis of protein structure. Protein structures must be in 3D conformation and can be downloaded from the Protein Data Bank (PDB). If a 3D conformation is not available, the structure can be obtained using homology modelling.
There must be no missing residues in the structure, and any that are missing must be added using the Modeller software. After the structure has been complexed with the best ligand, the input file for MD simulation must be prepared. A complex is formed in a system by adding water, after which the protein is minimised in a vacuum and a short MD is performed, restraining the protein. The system is then equilibrated and configured for the parameter being used. After all of the preparations have been completed, the long MD simulation can begin. 13 of the 21 articles reviewed are undergoing molecular dynamics testing to further validate the findings. Simulation times range from 1 ns to 250 ns, with an average of 50 ns to 100 ns. 11 of the 13 performed the simulation at 300 K, while only one performed the simulation at 310 K, and the other did not specify the simulation temperature.
Summary
Following our review of these articles, we made several recommendations to improve research in this area. To begin, it is critical to select target receptors of high quality for in silico experiments. This quality can be attributed to the structure having a lower resolution (below 2.5 Å) and no missing residues. This minor adjustment can significantly improve the quality of the research.
Second, a protein-protein interaction could help to ensure that the docking site is in the correct position. This allows the interacting residues of both receptors to be determined and provides additional evidence that the experiment is being carried out in the correct location.
Third, it is encouraged to use multiple receptor conformations for virtual screening and molecular docking. This can be accomplished by processing the same PDB file through MD simulation and capturing the conformation for each several ns. This gives virtual screening more consideration for what is matched with the receptors and what is not, while also semi-accommodating the protein's flexibility, which is non-rigid in nature. Another approach is to use multiple PDB files for the same receptor. This could provide insights into the consistency of the virtual screening results, such as whether the experiment used a different structure for the same macromolecule and still produced similar results.
Fourth, as discussed in the grid size section, it is recommended to set the grid size to the relative size of the ligands, which is 2.9 times larger than the radius of gyration of the docked molecule, according to the reference. As a result, when using a really big molecule for virtual screening and docking, this needs to be considered, because if it is too small as well, the search space becomes too narrow, making it ineffective.
Last but not least, molecular dynamics simulation should always be included in the research steps. This experiment could greatly aid the researcher's understanding of the ligand-receptor interaction, specifically how they interact with each other over time, which could be deduced as the stability of the ligand docked in the receptors. For each complex, 50 ns is a good starting point for the simulation duration, but longer simulations are preferable because they can provide more insights into the interaction. Trajectories analysis for MD simulation is also useful for research, and it is recommended that it be processed until the free energy calculation step using the MMGBSA/MMPBSA method.
Conclusions
We’ve shown several drug discovery research related to the ACE2 and SARS-CoV-2 Spike Protein by using the in-silico method. Aside from virtual screening, molecular docking, ADMET prediction, and molecular dynamic simulation, the research is carrying out several experiments. Several publications include active site determination, quantum chemical calculation, synthetic accessibility prediction, principal component analysis, and free energy calculation. These additional experiments could be extremely beneficial in ensuring the results of the lead compound or drug candidates. We've also highlighted the key interacting residues for each receptor reported in each article and summarised them in the order of how often they appeared in the reviewed articles. Finally, we've made several recommendations on how to make future research on this topic more elaborate and of higher quality, so that it can provide more precise results.
List of Abbreviations
Act. Site: Active Site Prediction; AD: AutoDock; ADMET: ADMET Prediction; ADT: AutoDockTools; ADV: AutoDockVina; AL: ADMET Lab; BSP: Binding Site Prediction; DFT: Density Functional Theory; DL: Drug-likeness; DSV: Discovery Studio Visualizer; DW: DataWarriot; FE: Free Energy Calculation; MD: Molecular Dynamics; Mi: Molinspiration Server; MOE: Molecular Operating Environment; MolDoc: Molecular Docking; PCA: Principal Component Analysis; PhyProp: Physical Properties; PISA: Protein Interface Stat. Analysis; PK: Pharmacokinetics; pkCSM: pkCSM Server; PLI: Protein-Ligand Interaction; PT2: ProTox-II; Qua. Calc. : Quantum Chemical Calculation; SA: SwissADME; SAP: Syntetic Accesibility Prediction; SP: Spike Protein; VS: Virtual Screening.
Declarations
Conflict of Interest
The authors declare no conflicting interest.
Data Availability
Not applicable.
Ethics Statement
Not applicable.
Funding Information
Not applicable.
References
- Fehr AR, Perlman S. Coronaviruses: an overview of their replication and pathogenesis. Methods Mol Biol [Internet]. 2015;1282:1–23. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25720466
- Weiss SR, Leibowitz JL. Coronavirus Pathogenesis. In: Advances in Virus Research [Internet]. 1st ed. Elsevier Inc.; 2011. p. 85–164. Available from: http://dx.doi.org/10.1016/B978-0-12-385885-6.00009-2
- Dries DJ. The Virus. Air Med J. 2020;39(4):231–4.
- Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N Engl J Med [Internet]. 2020 Feb 20;382(8):727–33. Available from: http://www.nejm.org/doi/10.1056/NEJMoa2001017
- Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell [Internet]. 2020;181(2):281-292.e6. Available from: http://dx.doi.org/10.1016/j.cell.2020.02.058
- Zhou P, Yang X Lou, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature [Internet]. 2020;579(7798):270–3. Available from: http://dx.doi.org/10.1038/s41586-020-2012-7
- Hinkson I V., Madej B, Stahlberg EA. Accelerating Therapeutics for Opportunities in Medicine: A Paradigm Shift in Drug Discovery. Front Pharmacol. 2020;11(June):1–7.
- Myers S, Baker A. Drug discovery - An operating model for a new era. Despite the advent of new science and technologies, drug developers will need to make radical changes in their operations if they are to remain competitive and innovative. Nat Biotechnol. 2001;19(8):727–30.
- Sun D, Gao W, Hu H, Zhou S. Why 90% of clinical drug development fails and how to improve it? Acta Pharm Sin B [Internet]. 2022;12(7):3049–62. Available from: https://doi.org/10.1016/j.apsb.2022.02.002
- Shaker B, Ahmad S, Lee J, Jung C, Na D. In silico methods and tools for drug discovery. Comput Biol Med [Internet]. 2021;137(July):104851. Available from: https://doi.org/10.1016/j.compbiomed.2021.104851
- Chen L, Morrow JK, Tran HT, Phatak SS, Du-Cuny L, Zhang S. From laptop to benchtop to bedside: structure-based drug design on protein targets. Curr Pharm Des [Internet]. 2012;18(9):1217–39. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22316152
- Ou-Yang SS, Lu JY, Kong XQ, Liang ZJ, Luo C, Jiang H. Computational drug discovery. Acta Pharmacol Sin. 2012;33(9):1131–40.
- Page MJ, McKenzie JE, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ. 2021;372.
- Mohamed K, Yazdanpanah N, Saghazadeh A, Rezaei N. Computational drug discovery and repurposing for the treatment of COVID-19: A systematic review. Bioorg Chem [Internet]. 2021;106(October 2020):104490. Available from: https://doi.org/10.1016/j.bioorg.2020.104490
- Aminpour M, Montemagno C, Tuszynski JA. An overview of molecular modeling for drug discovery with specific illustrative examples of applications. Molecules. 2019;24(9).
- Berendsen HJC, van der Spoel D, van Drunen R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput Phys Commun. 1995;91(1–3):43–56.
- Czaplewski C, Karczyńska A, Sieradzan AK, Liwo A. UNRES server for physics-based coarse-grained simulations and prediction of protein structure, dynamics and thermodynamics. Nucleic Acids Res. 2018;46(W1):W304–9.
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26(16):1781–802.
- D. E. Shaw Research. Desmond Molecular Dynamics System. Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY, 2021; 2022.
- Salomon-Ferrer R, Case DA, Walker RC. An overview of the Amber biomolecular simulation package. Wiley Interdiscip Rev Comput Mol Sci. 2013;3(2):198–210.
- Bahadur Gurung A, Ajmal Ali M, Elshikh MS, Aref I, Amina M, Lee J. An in silico approach unveils the potential of antiviral compounds in preclinical and clinical trials as SARS-CoV-2 omicron inhibitors. Saudi J Biol Sci [Internet]. 2022;29(6):103297. Available from: https://www.sciencedirect.com/science/article/pii/S1319562X22002133
- Kulkarni SA, Nagarajan SK, Ramesh V, Palaniyandi V, Selvam SP, Madhavan T. Computational evaluation of major components from plant essential oils as potent inhibitors of SARS-CoV-2 spike protein. J Mol Struct [Internet]. 2020;1221:128823. Available from: https://www.sciencedirect.com/science/article/pii/S0022286020311480
- Rameshkumar MR, Indu P, Arunagirinathan N, Venkatadri B, El-Serehy HA, Ahmad A. Computational selection of flavonoid compounds as inhibitors against SARS-CoV-2 main protease, RNA-dependent RNA polymerase and spike proteins: A molecular docking study. Saudi J Biol Sci [Internet]. 2021;28(1):448–58. Available from: https://doi.org/10.1016/j.sjbs.2020.10.028
- Natesh J, Mondal P, Penta D, Abdul Salam AA, Meeran SM. Culinary spice bioactives as potential therapeutics against SARS-CoV-2: Computational investigation. Comput Biol Med [Internet]. 2021;128:104102. Available from: https://www.sciencedirect.com/science/article/pii/S0010482520304339
- Al-Shuhaib MBS, Hashim HO, Al-Shuhaib JMB. Epicatechin is a promising novel inhibitor of SARS-CoV-2 entry by disrupting interactions between angiotensin-converting enzyme type 2 and the viral receptor binding domain: A computational/simulation study. Comput Biol Med [Internet]. 2022;141:105155. Available from: https://www.sciencedirect.com/science/article/pii/S0010482521009495
- Hadni H, Fitri A, Benjelloun AT, Benzakour M, Mcharfi M. Evaluation of flavonoids as potential inhibitors of the SARS-CoV-2 main protease and spike RBD: Molecular docking, ADMET evaluation and molecular dynamics simulations. J Indian Chem Soc [Internet]. 2022;99(10):100697. Available from: https://www.sciencedirect.com/science/article/pii/S0019452222003594
- Kar B, Dehury B, Singh MK, Pati S, Bhattacharya D. Identification of phytocompounds as newer antiviral drugs against COVID-19 through molecular docking and simulation based study. J Mol Graph Model [Internet]. 2022;114(April):108192. Available from: https://doi.org/10.1016/j.jmgm.2022.108192
- Vardhan S, Sahoo SK. In silico ADMET and molecular docking study on searching potential inhibitors from limonoids and triterpenoids for COVID-19. Comput Biol Med [Internet]. 2020;124:103936. Available from: https://www.sciencedirect.com/science/article/pii/S0010482520302729
- Kiran G, Karthik L, Shree Devi MS, Sathiyarajeswaran P, Kanakavalli K, Kumar KM, et al. In Silico computational screening of Kabasura Kudineer - Official Siddha Formulation and JACOM against SARS-CoV-2 spike protein. J Ayurveda Integr Med [Internet]. 2022;13(1):100324. Available from: https://www.sciencedirect.com/science/article/pii/S0975947620300243
- Jain AS, Sushma P, Dharmashekar C, Beelagi MS, Prasad SK, Shivamallu C, et al. In silico evaluation of flavonoids as effective antiviral agents on the spike glycoprotein of SARS-CoV-2. Saudi J Biol Sci [Internet]. 2021;28(1):1040–51. Available from: https://www.sciencedirect.com/science/article/pii/S1319562X20306136
- Khan AA, Baildya N, Dutta T, Ghosh NN. Inhibitory efficiency of potential drugs against SARS-CoV-2 by blocking human angiotensin converting enzyme-2: Virtual screening and molecular dynamics study. Microb Pathog [Internet]. 2021;152(July 2020):104762. Available from: https://www.sciencedirect.com/science/article/pii/S0882401021000346
- Benítez-Cardoza CG, Vique-Sánchez JL. Potential inhibitors of the interaction between ACE2 and SARS-CoV-2 (RBD), to develop a drug. Life Sci [Internet]. 2020;256:117970. Available from: https://www.sciencedirect.com/science/article/pii/S0024320520307207
- Gowrishankar S, Muthumanickam S, Kamaladevi A, Karthika C, Jothi R, Boomi P, et al. Promising phytochemicals of traditional Indian herbal steam inhalation therapy to combat COVID-19 – An in silico study. Food Chem Toxicol [Internet]. 2021;148:111966. Available from: https://www.sciencedirect.com/science/article/pii/S0278691520308565
- Muhseen ZT, Hameed AR, Al-Hasani HMHH, Tahir ul Qamar M, Li G. Promising terpenes as SARS-CoV-2 spike receptor-binding domain (RBD) attachment inhibitors to the human ACE2 receptor: Integrated computational approach. J Mol Liq [Internet]. 2020;320:114493. Available from: https://www.sciencedirect.com/science/article/pii/S0167732220359389
- Akinlalu AO, Chamundi A, Yakumbur DT, Afolayan FIDD, Duru IA, Arowosegbe MA, et al. Repurposing FDA-approved drugs against multiple proteins of SARS-CoV-2: An in silico study. Sci African [Internet]. 2021;13:e00845. Available from: https://doi.org/10.1016/j.sciaf.2021.e00845
- Baby K, Maity S, Mehta CH, Suresh A, Nayak UY, Nayak Y. SARS-CoV-2 entry inhibitors by dual targeting TMPRSS2 and ACE2: An in silico drug repurposing study. Eur J Pharmacol [Internet]. 2021;896:173922. Available from: https://www.sciencedirect.com/science/article/pii/S0014299921000753
- Pokhrel S, Bouback TA, Samad A, Nur SM, Alam R, Abdullah-Al-Mamun M, et al. Spike protein recognizer receptor ACE2 targeted identification of potential natural antiviral drug candidates against SARS-CoV-2. Int J Biol Macromol [Internet]. 2021;191(July):1114–25. Available from: https://www.sciencedirect.com/science/article/pii/S0141813021020754
- Yu R, Li P. Screening of potential spike glycoprotein / ACE2 dual antagonists against COVID-19 in silico molecular docking. J Virol Methods [Internet]. 2022;301:114424. Available from: https://www.sciencedirect.com/science/article/pii/S0166093421003633
- Singh P, Chauhan SS, Pandit S, Sinha M, Gupta S, Gupta A, et al. The dual role of phytochemicals on SARS-CoV-2 inhibition by targeting host and viral proteins. J Tradit Complement Med [Internet]. 2022;12(1):90–9. Available from: https://doi.org/10.1016/j.jtcme.2021.09.001
- Mishra GP, Bhadane RN, Panigrahi D, Amawi HA, Asbhy CR, Tiwari AK. The interaction of the bioflavonoids with five SARS-CoV-2 proteins targets: An in silico study. Comput Biol Med [Internet]. 2021;134(April):104464. Available from: https://doi.org/10.1016/j.compbiomed.2021.104464
- Nabati F, kamyabiamineh A, Kosari R, Ghasemi F, Seyedebrahimi S, Mohammadi S, et al. Virtual screening based on the structure of more than 105 compounds against four key proteins of SARS-CoV-2: MPro, SRBD, RdRp, and PLpro. Informatics Med Unlocked [Internet]. 2022;35(September):101134. Available from: https://www.sciencedirect.com/science/article/pii/S2352914822002714
- PDB-101. Resolution [Internet]. Available from: https://pdb101.rcsb.org/learn/guide-to-understanding-pdb-data/resolution
- Zhang H, Wang Y, Xu F. Impact of the subtle differences in MMP-12 structure on Glide-based molecular docking for pose prediction of inhibitors. J Mol Struct [Internet]. 2014;1076:153–9. Available from: http://dx.doi.org/10.1016/j.molstruc.2014.06.002
- Stanzione F, Giangreco I, Cole JC. Use of molecular docking computational tools in drug discovery [Internet]. 1st ed. Vol. 60, Progress in Medicinal Chemistry. Elsevier B.V.; 2021. 273–343 p. Available from: http://dx.doi.org/10.1016/bs.pmch.2021.01.004
- Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem [Internet]. 2009 Dec;30(16):2785–91. Available from: https://onlinelibrary.wiley.com/doi/10.1002/jcc.21256
- Trott O, Olson AJ. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem [Internet]. 2009;NA-NA. Available from: https://onlinelibrary.wiley.com/doi/10.1002/jcc.21334
- Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, et al. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J Med Chem [Internet]. 2006 Oct 1;49(21):6177–96. Available from: https://pubs.acs.org/doi/10.1021/jm051256o
- Cresset®. Flare [Internet]. Litlington, Cambridgeshire, UK; Available from: http://www.cresset-group.com/flare/
- Chemical Computing Group ULC. Molecular Operating Environment (MOE). 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7: 2022.02; 2022.
- National Center of Biotechnology Information. PubChem [Internet]. 2023. Available from: https://pubchem.ncbi.nlm.nih.gov/
- Wishart DS, Feunang YD, Guo AC, Lo EJ, Marcu A, Grant JR, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res [Internet]. 2018 Jan 4;46(D1):D1074–82. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29126136
- Green Pharma. Ambinter [Internet]. Available from: https://ambinter.com/#search
- ChemDiv. ChemDiv [Internet]. Available from: https://www.chemdiv.com/
- Patrick Walters W, Stahl MT, Murcko MA. Virtual screening - An overview. Drug Discov Today. 1998;3(4):160–78.
- Meyer-Almes FJ. Repurposing approved drugs as potential inhibitors of 3CL-protease of SARS-CoV-2: Virtual screening and structure based drug design. Comput Biol Chem [Internet]. 2020;88(June):107351. Available from: https://doi.org/10.1016/j.compbiolchem.2020.107351
- van Drie JH. Computer-aided drug design: The next 20 years. J Comput Aided Mol Des. 2007;21(10–11):591–601.
- van Drie JH. Pharmacophore discovery--lessons learned. Curr Pharm Des [Internet]. 2003;9(20):1649–64. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12871063
- Mysinger MM, Carchia M, Irwin JJ, Shoichet BK. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J Med Chem. 2012;55(14):6582–94.
- Gaulton A, Hersey A, Nowotka M, Bento AP, Chambers J, Mendez D, et al. The ChEMBL database in 2017. Nucleic Acids Res [Internet]. 2017 Jan 4;45(D1):D945–54. Available from: https://academic.oup.com/nar/article-lookup/doi/10.1093/nar/gkw1074
- Tripathi A, Misra K. Molecular Docking: A Structure-Based Drug Designing Approach. JSM Chem. 2017;5(2):1042.
- Davis IW, Baker D. RosettaLigand docking with full ligand and receptor flexibility. J Mol Biol [Internet]. 2009 Jan 16;385(2):381–92. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19041878
- Jain AN. Surflex: Fully Automatic Flexible Molecular Docking Using a Molecular Similarity-Based Search Engine. J Med Chem [Internet]. 2003 Feb 1;46(4):499–511. Available from: https://pubs.acs.org/doi/10.1021/jm020406h
- Venkatachalam CM, Jiang X, Oldfield T, Waldman M. LigandFit: a novel method for the shape-directed rapid docking of ligands to protein active sites. J Mol Graph Model [Internet]. 2003 Jan;21(4):289–307. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12479928
- Kumar S, Kumar S. Molecular Docking: A Structure-Based Approach for Drug Repurposing [Internet]. In Silico Drug Design: Repurposing Techniques and Methodologies. Elsevier Inc.; 2019. 161–189 p. Available from: http://dx.doi.org/10.1016/B978-0-12-816125-8.00006-7
- Meng X-Y, Zhang H-X, Mezei M, Cui M. Molecular Docking: A Powerful Approach for Structure-Based Drug Discovery. Curr Comput Aided-Drug Des. 2011;7(2):146–57.
- Ferreira LG, Dos Santos RN, Oliva G, Andricopulo AD. Molecular docking and structure-based drug design strategies. Vol. 20, Molecules. 2015. 13384–13421 p.
- Ma DL, Chan DSH, Leung CH. Drug repositioning by structure-based virtual screening. Chem Soc Rev. 2013;42(5):2130–41.
- Feinstein WP, Brylinski M. Calculating an optimal box size for ligand docking and virtual screening against experimental and predicted binding pockets. J Cheminform [Internet]. 2015;7(1). Available from: http://dx.doi.org/10.1186/s13321-015-0067-5
- Towler P, Staker B, Prasad SG, Menon S, Tang J, Parsons T, et al. ACE2 X-Ray Structures Reveal a Large Hinge-bending Motion Important for Inhibitor Binding and Catalysis. J Biol Chem [Internet]. 2004 Apr;279(17):17996–8007. Available from: http://www.nature.com/articles/nsb1203-980
- Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581(7807):215–20.
- Perricone U, Gulotta MR, Lombino J, Parrino B, Cascioferro S, Diana P, et al. An overview of recent molecular dynamics applications as medicinal chemistry tools for the undruggable site challenge. Medchemcomm. 2018;9(6):920–36.