RESEARCH ARTICLE
In Silico Structure-Activity Study of Selected Triterpenoids as Potential Inhibitors of Mycolic Acid Transporter of Mycobacterial MmpL3 Receptor Protein
Sciences of Phytochemistry|Vol. 3, Issue 2, pp. 82-90 (2024)
CC BY 4.0-2024 Authors
Views
Downloads
Shares
Received
Apr 25, 2024Revised
Jun 24, 2024Accepted
Aug 1, 2024Published
Sep 5, 2024
Abstract
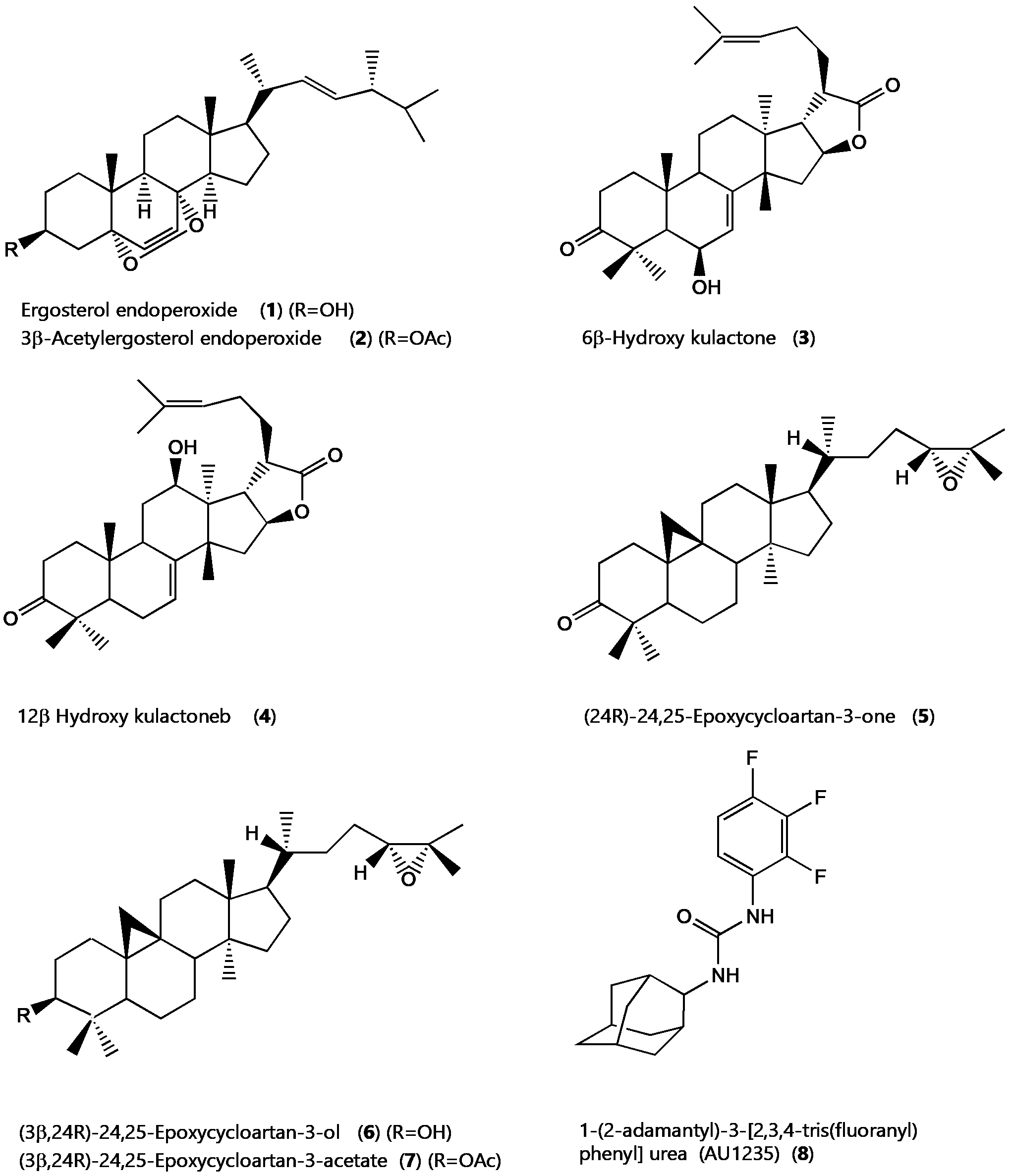
Structural features of the triterpenoid skeleton that are necessary for antimycobacterial activity are not well understood. Following the isolation of the triterpenoids ergosterol-5,8-endoperoxide, 6β-hydroxykulactone, 12β-hydroxykulactone, (24R)-24,25-epoxycycloartan-3-one and (3β,24R)-24,25-epoxycycloartan-3-ol, and (3β,24R)-24,25-epoxycycloartan-3-acetate with varying antimycobacterial activity ranging between MIC of 1 µg/ml to128 µg/ml prompted us to study this class of compounds further to shade light on the structural features necessary for their antimycobacterial activity. This in silico study involved docking the triterpenoids on the mycobacterial multi-pharmacophore receptor protein MmpL3. The docking results were compared with the MmpL3 receptor protein co-crystallized TB drug candidate, AU1235, (1-(2-adamantyl)-3-[2,3,4-tris(fluoranyl)phenyl] urea). The virtual screening revealed key structural features in the triterpenoid skeleton, including the C-3 keto and β-hydroxy group on C-3 or C-6, as important for antimycobacterial activity. Also, the decreased binding affinity for compounds 2 and 7 with an acetate group on C-3 were in tandem with those observed in vitro. Toxicity predictions revealed that this class of compounds had no mutagenic effects and displayed favorable pharmacokinetic parameters. The study reveals the potential of the triterpenoid skeleton exemplified by the readily available ergosterol-5,8-endoperoxide as a useful scaffold in searching and developing effective therapeutic lead entities to facilitate anti-tuberculosis drug discovery.
Introduction
Mycobacterium tuberculosis (Mtb) is the causative agent of tuberculosis (TB), an infectious disease that continues to be a threat to global public health, especially in low-to-middle-income countries (1). TB is a preventable and usually curable disease. Yet in 2022, TB was the world's second leading cause of death from a single infectious agent, after coronavirus disease (COVID-19), and caused almost twice as many deaths as HIV/AIDS. TB was responsible for approximately 1.3 million deaths in 2022 (2, 3). With the emergence of multidrug-resistant tuberculosis (MDR-TB), extensively drug-resistant tuberculosis (XDR-TB), and the resistance of multiple first-line and second-line TB medications, including isoniazid and ethionamide, novel therapeutic agents are urgently needed, and continued drug discovery efforts required (4, 5).
Identifying novel receptor proteins as targets of a broad number of compounds with varying chemical scaffolds has provided essential tools to understand the mode of action of potential antimycobacterial compounds at the Mtb protein receptor level (6, 7). MmpLs (Mycobacterial membrane proteins Large), which play crucial roles in transporting lipids, polymers, and immunomodulators and extrude therapeutic drugs, have been among the most important therapeutic drug targets to emerge recently (8). MmpL3, one of these proteins, is an essential mycolic acid and lipid transporter required for growth cell viability and is crucial for cell-wall biosynthesis (9, 10, 11). More than thirty pharmacophores have been reported to target MmpL3 (12, 13). Since MmpL3 is involved in key pathways for M. tuberculosis survival, it could be exploited to develop novel anti-TB drugs (9, 10). These developments prompted us to re-evaluate the antimycobacterial terpenoids we isolated earlier and co-workers (14-17) using in silico techniques, including molecular docking.
In this study, seven triterpenoids (see Figure 1) were docked into the ligand-binding pocket of the multi-pharmacophore MmpL3 receptor protein (PDP ID: 6ajh) to investigate their interactions with the amino acids in the selected pocket. The binding pocket was identified based on the protein-inhibitor interactions observed in the MmpL3 complex with the inhibitor AU1235 (1-(2-adamantyl)-3-[2, 3, 4-tris (fluoranyl)phenyl] urea). Additionally, the binding affinities of the triterpenoids were compared with those of the TB drug candidate AU1235. The insights gained from the ligand-receptor interactions within the binding site will enhance our understanding of the structural features in the terpenoid skeleton necessary for their observed antimycobacterial activity. To assess the in vivo potential of these compounds as anti-TB candidates, an ADMET (absorption, distribution, metabolism, excretion, toxicity) profile evaluation was conducted (18, 19). The results were expected to support earlier in vitro studies and demonstrate the value of in silico approaches in understanding the modes of action of the selected triterpenoids, offering opportunities to develop novel antimycobacterial agents.
Experimental Section
Molecular docking was carried out using Autodock Vina embedded in PyRx 0.8. The receptor protein MmpL3 (PDB ID: 6ajh; 2.82 A) was employed as the receptor protein (20). The selected triterpenoids were used as ligands.
Ligand Retrieval and Preparation
The selection of the ligand molecules was based on earlier studies on the antimycobacterial activity of two East African medicinal plants, Ajuga remota, and Melia volkensii, coupled with a literature survey. Seven triterpenoids, compounds 1-7 (see Figure 1), whose antimycobacterial potential was investigated in vitro earlier, were selected for the current in silico study (14, 15, 17). The first-line anti-TB drug isoniazid was used as a comparative standard (21). The MmpL3 inhibitor or AU1235, 1-(2-adamantyl)-3-[2, 3, 4-tris (fluoranyl) phenyl]urea, was used to validate the docking exercise (22). 3D structures of compounds to be studied were downloaded in SDF format from the PubChem database (https://pubchem. ncbi. nlm. nih. gov/) (1-6) is a public repository for information on chemical substances and their biological activities. The compounds were then energy minimized by Open Babel 2.3.1 using the MMFF94 force field with an energy gradient of 0.05 using the steepest descent algorithm, 500 numbers steps, and 10e-7 convergence parameter to generate an energetically reasonable posture for docking. The various optimized 3D structures were finalized for docking using Autodock tools 1.5.7 and saved as PDBQT files for the subsequent steps.
Receptor Protein and Molecular Docking Preparation
The MmpL3 receptor protein (PDB ID: 6ajh) was selected based on information from the literature (9). The 3D structure of the receptor protein in complex with AU1235 (1-(2-adamantyl)-3-[2, 3, 4-tris (fluoranyl)phenyl]urea) was obtained from the Protein Data Bank repository (https://www. rcsb. org; accessed on March 3rd, 2024) (20). The receptor protein was prepared for docking using UCSF ChimeraX (Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco) and AutoDock Tools 1.5.7 (Scripps Research, La Jolla, California, USA). The preparation involved removing unwanted water molecules, bound heteroatoms, and other ligands. Polar hydrogens and Kollman charges were added, and the protein molecule was saved in. pdbqt format.
Docking studies for the target protein (PDB ID: 6ajh) and various triterpenoid ligands were performed using AutoDock Vina, embedded within PyRx 0.8, and the results were visualized with PyMOL, Protein Plus, and Discovery Studio Visualizer version 2020. The grid box was centered on the binding pocket with Vina search space parameters: Center (Å) x: 35.8727, y: 3.9155, z: 20.7025; Dimensions (Å) x: 23.4367, y: 22.1532, z: 22.1676. An exhaustiveness level of 8 was used. The binding interactions between the protein and ligands were analyzed and visualized using Discovery Studio Visualizer version 4.0. The docking results were then clustered based on root mean square deviation (RMSD) to assess the structural variability of the binding conformations. The clusters were ranked according to the predicted free energy of binding, which provides an estimation of the strength and stability of each protein-ligand interaction. Binding postures were further analyzed using Ligplot Plus version 2.2.8 and Protein Plus to evaluate the specific contacts and interactions, such as hydrogen bonds and hydrophobic interactions. Finally, the dissociation constants (Kd) were calculated using the online tool NovoPro at www. novoprolabs. com/tools/deltag2kd.
Docking Validation
To validate the docking process, the native ligand co-crystallised with the protein was extracted and redocked in the corresponding binding pocket of the receptor protein. The ligand's ability to reproduce the ligand's orientation and position in the bound form was determined using the Root Mean Square Deviation (RMSD) of the superimposed co-crystal and posting pose of the native ligand.
Results and Discussion
Isolation of Compounds
During a bioassay-guided study reported earlier, Ajuga remota led to the isolation of ergosterol-5, 8-endoperoxide compound 1 with a MIC of 1mg/mL. Acetylation of compound 1 gave the acetate compound 2, with an MIC of 8mg/mL (14, 16). Studies on Melia volkensii resulted in isolating 12β-hydroxykulactone compound 3 and 6β-hydroxykulactone compound 4. Compound 3 had an MIC of 16mg/mL, while compound 4 had an MIC of 4mg/mL (15, 16). Studies on Borrichia frutescens by co-workers afforded (24R)-24, 25-epoxycycloartan-3-one compound 5 and (3β, 24R)-24, 25-epoxycycloartan-3ol compound 6 with MICs of 8mg/mL, whereas compound 7 the acetate of compound 6 was inactive with an MIC > 128mg/mL (16, 17). Correlations of structural features and the MICs of the seven triterpenoid compounds 1–7 suggest that the presence of the C-3 keto or β-hydroxy group, the cyclopropane ring, and the epoxide moieties, as well as the presence of a β-hydroxyl group on C-6 or C-12, seem to play an important role in the observed in vitro anti-tuberculosis activity of these compounds. However, the apparent importance of these functional groups is lacking structural evidence at the relevant protein receptor level. Further studies utilizing crystallography or cryo-EM are needed to elucidate the precise binding interactions of these triterpenoids with their target proteins.
| Ligand | In vitro MIC µg/ml | Binding affinity kcal/mol | Number of various interactions | ||||
|---|---|---|---|---|---|---|---|
| Alkyl | Pi-donor bond | Conv. H bonds | Pi- Sigmabond | Steric clash | |||
| Ergosterol endoperoxide | 1 | -10.8 | 4 | 1 | 0 | 0 | 0 |
| 3β-Acetoxy ergosteryl endoperoxide | 8 | -6.4 | 6 | 0 | 0 | 0 | 1 |
| 6β-Hydroxy kulactone | 4 | -7.3 | 5 | 1 | 0 | 1 | 1 |
| 12β-Hydroxy kulactone | 16 | -5.9 | 4 | 1 | 1 | 1 | 1 |
| (24R)-24, 25-epoxycycloartan-3-one | 8 | -6.4 | 0 | 2 | 0 | 0 | 1 |
| (3β, 24R)-24, 25-epoxycycloartan-3-ol | 8 | -7.8 | 6 | 0 | 0 | 1 | 1 |
| (3b, 24R)-24, 25-epoxycycloartan-3-acetate | > 128 | -4.3 | 4 | 0 | 0 | 0 | 2 |
| (1-(2-adamantyl)-3-[2, 3, 4-tris (fluoranyl)phenyl] urea) | - | -12.0 | 6 | 0 | 1 | 1 | 0 |
Molecular Docking
In the present study, seven triterpenoids with varying antimycobacterial tuberculosis activity were subjected to an in silico molecular docking on the multi-pharmacophore receptor protein MmpL3, an essential mycolic acid and lipid transporter ligand binding domain (LBD). The LBD is usually involved in receptor dimerization, ligand recognition, and cofactor protein interactions. A docking study was performed to evaluate the binding potential of the seven selected triterpenoids. The docking affinities calculated by Autodock Vina ranged between -4.5 to 10.5 kcal/mol and indicated strong binding to the target protein (see Table 1). This was compared with the anti-TB drug candidate AU1235 and the anti-TB drug isoniazid. From the docking study, ergosterol endoperoxide had a binding affinity of -10.8 kcal/mol, while ergosterol-3β-acetate endoperoxide had a lower binding affinity score of -6.4 kcal/mol. Also, (3β, 24R)-24, 25-epoxycycloartan-3-ol had a binding a binding score of -7.8 kcal/mol while (3β, 24R)-24, 25-epoxycycloartan-3-acetate had much a lower binding score of -4.3 kcal/mol. This observation is in tandem with the trend observed in the experimental in vitro results obtained earlier.
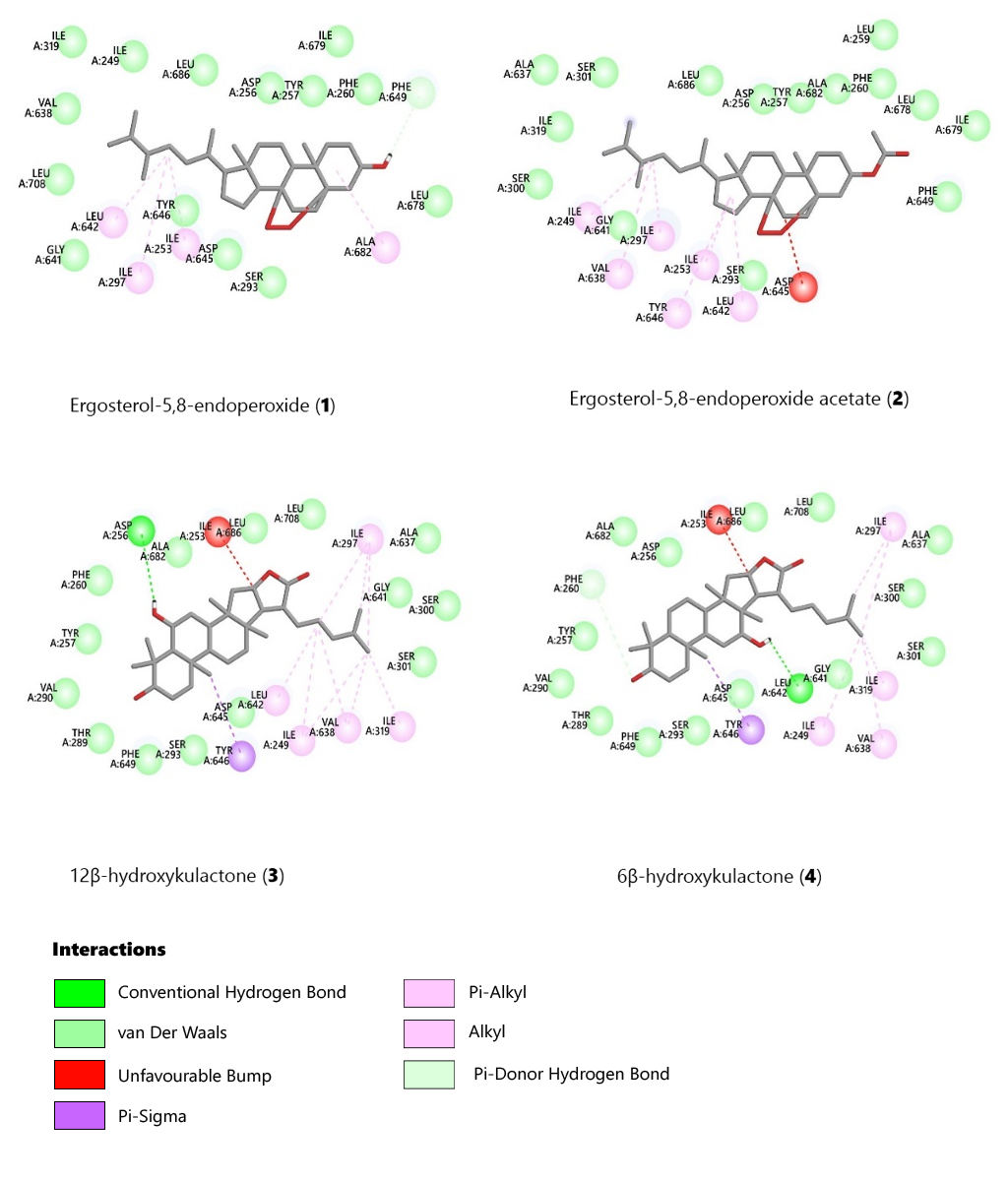
Figures 3 and 4 present docking interactions between the selected ligands and the receptor protein. Compounds 1 and 2 exhibited van der Waals interactions with Ser293, Gly641, Ile319, Leu686, Asp256, Tyr257, Phe260, Leu678, and Ile679. Both compounds also showed pi-alkyl and alkyl interactions with Ile253 and Ile297. However, compound 1 formed a pi-donor hydrogen bond between its 3β-hydroxy group and the amino acids Phe649 and Phe260, absent in the 3β-acetyl derivative (compound 2) (see Figure 2). The closely related derivatives (3β, 24R)-24, 25-epoxycycloartan-3-one (compound 5), (3β, 24R)-24, 25-epoxycycloartan-3-ol (compound 6), and (3β, 24R)-24, 25-epoxycycloartan-3-acetate (compound 7) also showed van der Waals interactions with similar amino acids, including Ser293, Ile679, Thr289, Tyr257, Tyr301, Asp256, and Ala682.
Compounds 6 and 7 shared alkyl interactions with Ile297, Ile249, and Val638, whereas compound 5 did not. Compounds 5 and 6 formed a pi-sigma bond with Phe649, while compound 7 did not. Additionally, compound 5 formed a pi-donor hydrogen bond between its 3-keto group and Phe260 (Figures 3 and 4). These docking results align with the observed in vitro antimycobacterial activity of compounds 5, 6, and 7, highlighting the significance of a hydroxyl or keto group at position C-3 of the epoxycycloartan skeleton.
Compounds 3 (12β-hydroxykulactone) and 4 (6β-hydroxykulactone) displayed the same van der Waals interactions with the amino acids Ser301, Ser300, Gly641, Ala637, Leu708, Leu686, Ala682, Asp256, Phe260, Tyr257, Val290, Thr289, Phe649, Ser293, and Asp645. The C-10 methyl group in compounds 3 and 4 formed a pi-sigma bond with Tyr646. However, compound 3 formed a conventional hydrogen bond with Asp256, while compound 4 formed one with Leu642. The docking results for compounds 3 and 4 support the observed in vitro antimycobacterial activity, emphasizing the importance of a β-hydroxyl group at positions C-6 and C-12.
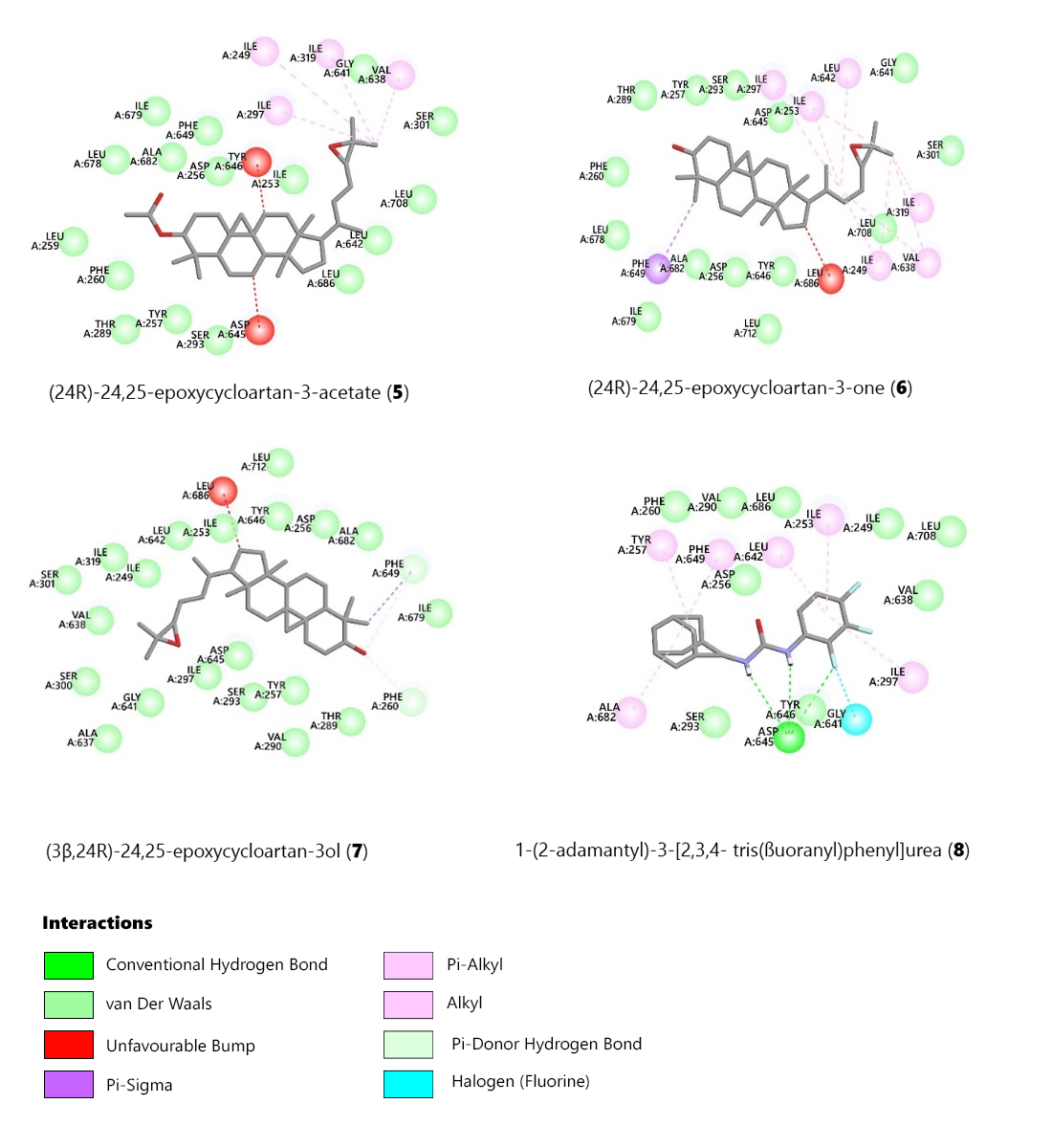
The C-4 methyl group in compounds 5 and 6 formed a pi-sigma bond with Phe649. In contrast, the 3-acetate derivative of compound 6 did not interact with the C-4 methyl groups. The best-docked triterpenoid conformers were selected based on their binding energy and favorable interactions with the amino acid residues in the active site.
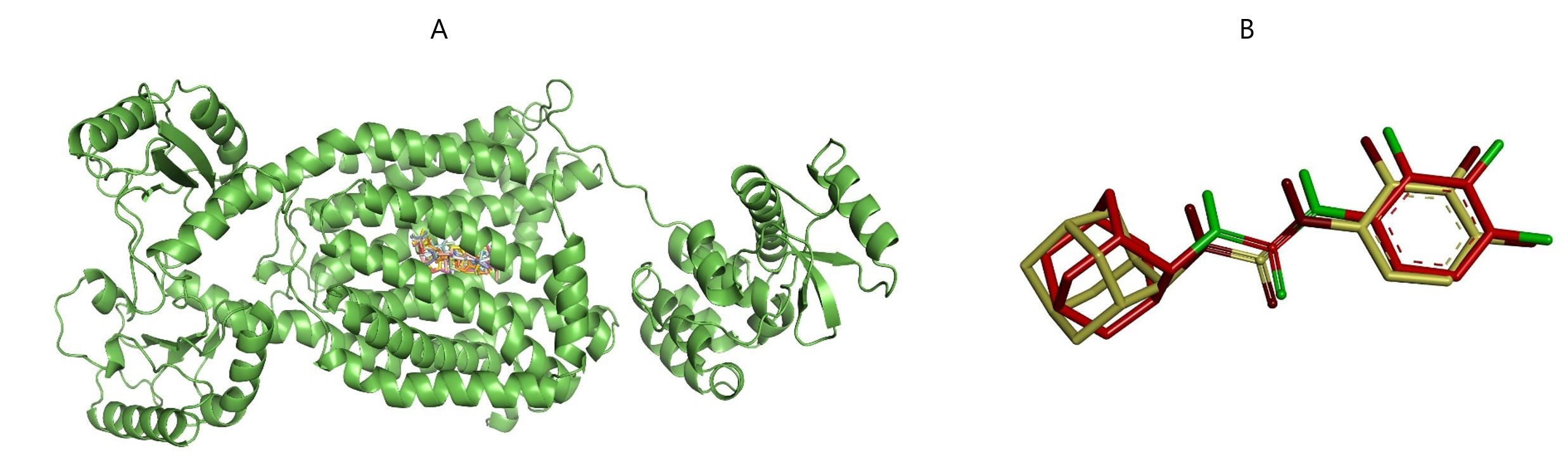
The MmpL3 receptor protein bound to compounds 1-7 at the native ligand AU1235 binding pocket is shown in Figure 2A. The binding energies and the various ligand-receptor amino acid interactions are listed in Table 1. The molecular docking procedure was validated by the ability of the native ligand to reproduce the pose and the position in the bound form, as shown in the superimposed co-crystal and post-docking pose of the native ligand depicted in Figure 2B. The Root Mean Square Deviation (RMSD) of the superimposed co-crystal and post-docking pose of the native ligand AU1235 was determined to be 1.2Å, an RMSD value < 2Å validating the docking procedure.
ADMET Prediction
ADMET is an abbreviation for Absorption, Distribution, Metabolism, Excretion, and Toxicity and plays key roles in drug discovery and development. A high-quality drug candidate should have sufficient efficacy against the therapeutic target and show appropriate ADMET properties.
| Ligand | Mwt | No. RBs | logP | Ro5 | LD50mg/kg | Carcinogen | Ames test | HIA | HBA | HBD |
|---|---|---|---|---|---|---|---|---|---|---|
| Ergosterol-5, 8-endoperoxide | 428.6 | 4 | 5.59 | 1 | 2340 | inactive | Non-mutagen | 0.96 | 3 | 1 |
| Ergosteryl -3β-acetate-5, 8-endoperoxide | 470.6 | 6 | 5.89 | 1 | 79 | inactive | Non-mutagen | 0.97 | 4 | 0 |
| 6β-Hydroxy kulactone | 468.6 | 3 | 5.31 | 1 | 914 | active | Non-mutagen | 0.96 | 4 | 1 |
| 12β-Hydroxy kulactone | 468.6 | 3 | 5, 24 | 1 | 2589 | active | Non-mutagen | 0.96 | 4 | 1 |
| (24R)-24, 25-epoxycycloartan-3-one | 440.7 | 4 | 6.27 | 1 | 16000 | inactive | Non-mutagen | 1.00 | 2 | 0 |
| (24R)-24, 25-epoxycycloartan-3-ol | 442.7 | 4 | 6.26 | 1 | 5000 | inactive | Non-mutagen | 0.97 | 2 | 1 |
| (24R)-24, 25-epoxycycloartan-3-acetate | 484.7 | 6 | 6.56 | 1 | 5000 | inactive | Non-mutagen | 0.98 | 3 | 0 |
| AU1235 | 324.3 | 4 | 3.42 | 0 | 4000 | inactive | Mutagen | 0.92 | 3 | 2 |
The underlying goal is to understand better a compound's toxicity and safety profile to decide whether the compound can be subjected to further studies. The five properties in ADMET can be determined experimentally and, to some degree, computationally (23). ADMET prediction was performed on the seven triterpenoids and AU1235. This was accomplished using Discovery Studio 4.5 (Accelrys Inc. , San Diego, CA, USA) and an online server PreADMET ver 2.0 (https://preadmet. bmdrc. kr) (24). The predicted parameters included human intestinal absorption (HIA), LogP, LD50, carcinogenicity, and Ames mutagenicity. From the ADMET data, the triterpenoids did not violate more than one Lepinski's rule of 5 (Ro5) and can, therefore, be categorized as drug-like compounds (25, 26). Also, they are safe with acceptable LD50 values, bioavailable, and non-mutagenic (Table 2).
Cell permeable compounds should have HBD (hydrogen bond donors) ≤ 5, HBA (hydrogen bond acceptors) ≤ 10), Mwt ≤ 500, and log P ≤ 5. No. RBs (number of rotatable bonds) should be ≤ 5. HIA (human intestinal absorption) bioavailability endpoint should be ≥ 0.5. Ro5 (Lepinski's rule of 5) should be ≤ 1.
Conclusions
The in silico study revealed that the β-hydroxy group on C-3 or C-6 are vital structural features in the triterpenoid skeleton for antimycobacterial activity. Toxicity predictions revealed that this class of compounds had no mutagenic effects and displayed favorable pharmacokinetic parameters. From these studies, it can be concluded that in silico techniques can be utilized to fast-track the development of new drugs to treat tuberculosis.
Overall, the study reveals the potential of the triterpenoid skeleton exemplified by the readily available ergosterol-5, 8-endoperoxide compound 1 as a helpful scaffold in searching for and developing effective therapeutic lead entities to facilitate anti-tuberculosis drug discovery.
Declarations
Acknowledgment
I wish to acknowledge the Pwani University Council for granting me sabbatical leave after my term as Vice Chancellor. I also wish to acknowledge the Technical University of Mombasa for hosting me as a Visiting Professor. The author is currently the Chairman of the Kenya National Commission for UNESCO (KNATCO).
Conflict of Interest
The author declares no conflicting interest.
Data Availability
The unpublished data is available upon request to the corresponding author.
Ethics Statement
Not applicable.
Funding Information
Not applicable.
References
- Goletti D, Al-Abri S, Migliori GB, Arlehamn CL, Haldar P, Sundling C, da Costa C, To KW, Martineau AR, Petersen E, Zumla A, Shui Shan Lee SS. World Tuberculosis Day 2024 theme "Yes! We can end TB" can be made a reality through concerted global efforts that advance detection, diagnosis, and treatment of tuberculosis infection and disease. Int J Infect Dis. 2024; 141: 1-3.
- World Health Organization. Global tuberculosis report 2023. Geneva: World Health Organization; 2023. Available from: https://iris. who. int/handle/10665/363752
- Glaziou P, Arinaminpathy N, Dodd PJ, Dean A, Floyd K. Methods used by WHO to estimate the global burden of TB disease 2023. Geneva: World Health Organization; 2022. Available from: https://www. who. int/publications/m/item/methods-used-by-who-to-estimate-the-global-burden-of-tb-disease
- Lechartier B, Rybniker J, Zumla A, Cole ST. Tuberculosis drug discovery in the post-post-genomic era. EMBO Mol Med. 2014; 6: 158– 68.
- Chiarelli LR, Mori G, Esposito M, Orena BS, Pasca MR. New and old hot drug targets in tuberculosis. Curr Med Chem. 2016; 23: 3813– 46.
- Kumar M, Singh SK, Singh PP, Singh VK, Rai AC, Srivastava AK, et al. Potential Anti-Mycobacterium tuberculosis Activity of Plant Secondary Metabolites: Insight with Molecular Docking Interactions. Antioxidants (Basel). 2021; 10: 2-25.
- Choksi H, Carbone J, Paradis NJ, Bennett L, Bui-Linh C, Chun Wu C. Novel Inhibitors to MmpL3 Transporter of Mycobacterium tuberculosis by Structure-Based High-Throughput Virtual Screening and Molecular Dynamics Simulations. ACS Omega. 2024; 9 (12): 13782-9.
- Schwartz EJ, Zgurskaya CP, Jackson HI. Recent advances in mycobacterial membrane protein large 3 inhibitor drug design for mycobacterial infections. Expert Opin Drug Discov. 2023; 18 (7): 707-24.
- Degiacomi G, Belardinelli JM, Pasca MR, De Rossi E, Riccardi G, Chiarelli LR. Promiscuous Targets for Antitubercular Drug Discovery: The Paradigm of DprE1 and MmpL3. Appl Sci. 2020; 10: 2-18.
- Li W, Upadhyay A, Fontes FL, North EJ, Wang Y, Crans DC, et al. Novel insights into the mechanism of inhibition of MmpL3, a target of multiple pharmacophores in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2014; 58: 6413– 23.
- Lee BS, Pethe K. Therapeutic potential of promiscuous targets in Mycobacterium tuberculosis. Curr Opin Pharmacol. 2018; 42: 22– 6.
- Williams JT, Abramovitch RB. Molecular Mechanisms of MmpL3 Function and Inhibition. Microb Drug Resist. 2023; 29 (5): 190-2012.
- Grover S, Engelhart CA, Perez-Herran E. Two-way regulation of MmpL3 expression identifies and validates inhibitors of MmpL3 function in Mycobacterium tuberculosis. ACS Infect Dis. 2021; 7 (1): 141– 52.
- Cantrell CL, Rajab MS, Franzblau SG, Fronczek FR, Fischer NH. Antimycobacterial ergosterol-5, 8-endoperoxide from Ajuga remota. Planta Med. 1999; 65: 732– 4.
- Cantrell CL, Rajab MS, Franzblau SG, Fischer NH. Antimycobacterial triterpenes from Melia volkensii. J Nat Prod. 1999; 62: 546-8.
- Cantrell CL, Franzblau SG, Fisher NH. Antimycobacterial plant terpenoids. Planta Med. 2001; 67: 685– 94.
- Cantrell CL, Lu T, Fronczek FR, Fischer NH, Adams LB, Franzblau SG. Antimycobacterial cycloartanes from Borrichia frutescens. J Nat Prod. 1996; 59: 1131-6.
- Ló pez-Nieto B, Estrada-Tejedor R, Borrell JI. Evaluation of Free Online ADMET Tools for Academic or Small Biotech Environments. Molecules. 2023; 28 (2): 776.
- Xiong G, Wu Z, Yi J, Fu L, Yang Z, Hsieh C, et al. ADMETlab 2.0: An Integrated Online Platform for Accurate and Comprehensive Predictions of ADMET Properties. Nucleic Acids Res. 2021; 49
- – W14.
- Zhang B, Li J, Yang X, Wu L, Zhang J, Yang Y, et al. Crystal Structures of Membrane Transporter MmpL3, an Anti-TB Drug Target. Cell. 2019; 176: 636-48.
- Khan SR, Manialawy Y, Siraki AG. Isoniazid and host immune system interactions: A proposal for a novel comprehensive mode of action. Br J Pharmacol. 2019; 176: 4599– 608.
- Bell EW, Zhang Y. DockRMSD: an open-source tool for atom mapping and RMSD calculation of symmetric molecules through graph isomorphisim. J Cheminform. 2019; 11: 40.
- Lipinski CA. ADMET Screen. In: Schwab M, editor. Encyclopedia of Cancer. Berlin: Springer; 2011. https://doi. org/10.1007/978-3-642-16483-5_114.
- Knoll KE, van der Walt MM, Lootsa DT. In Silico Drug Discovery Strategies Identified ADMET Properties of Decoquinate RMB041 and Its Potential Drug Targets against Mycobacterium tuberculosis. Microbiol Spectr. 2022; 10 (2): 1-17.
- Lipinski CA. Lead and drug-like compounds: the rule-of-five revolution. Drug Discov Today Technol. 2004; 1 (4): 337– 41.
- Maliehe TS, Tsilo PH, Shandu JS. Computational Evaluation of ADMET Properties and Bioactive Score of Compounds from Encephalartos ferox. Pharmacogn J. 2020; 12 (6): 1357-62.