RESEARCH ARTICLE
Druggability of Pharmaceutical Compounds Using Lipinski Rules with Machine Learning
Academic Editor: Ernest Domanaanmwi Ganaa
Sciences of Pharmacy|Vol. 3, Issue 4, pp. 177-192 (2024)
CC BY 4.0-2024 Authors
Views
Downloads
Shares
Received
Jul 17, 2024Revised
Oct 18, 2024Accepted
Nov 9, 2024Published
Nov 11, 2024
Abstract
In the field of pharmaceutical research, identifying promising pharmaceutical compounds is a critical challenge. The observance of Lipinski's Rule of Five (RO5) is a fundamental criterion, but evaluating many compounds manually requires significant resources and time. However, the integration of computational techniques in drug discovery in its early stages has significantly transformed the pharmaceutical industry, enabling further efficient screening and selection of possible drug candidates. Therefore, this study explores RO5 using algorithms of Machine Learning (ML), offering a comprehensive method to predict the druggability of pharmaceutical compounds. The study developed, evaluated, and validated the performance metrics of multiple supervised machine learning models. The best model was used to build an application that can predict and classify potential drug candidates. The findings revealed promising capabilities across all models for drug classification. Among all the explored models, Random Forest (RF), Extreme Gradient Boost (XGBoost), and Decision Tree (DT) classifiers demonstrated exceptional performance, achieving near-perfect accuracy of 99.94%, 99.81% and 99.87% respectively. This highlights the robustness of ensemble learning methods in classifying compounds based on RO5 adherence. The comparative analysis of these models underscores the importance of considering balanced accuracy, precision, F1-score, recall, and Receiver Operating Characteristics-Area Under the Curve (ROC-AUC) score, interpretability, and computational efficiency when choosing between ML algorithms in drug discovery. The DrugCheckMaster application was subsequently developed using the most predictive model and is now available on Render (https://capstone-project-dc7w. onrender. com/).
Introduction
The process of drug discovery includes identifying drug candidates, synthesizing, characterizing, and screening them for therapeutic efficacy (1). It includes target discovery, lead discovery, lead optimization, preclinical development, three phases of clinical trials, and lastly, market launch, provided all regulations are met (2). However, this developmental process is one of the most challenging human applications, as it demands a delicate balance between ensuring safety within an appropriate therapeutic range and maximizing efficacy in delivering health benefits (3). The likelihood of drug candidates successfully advancing through Phase I clinical development is estimated to be only 7–11% (4). With so many challenges inherent in drug development particularly the drug candidates’ low success rate of advancing to clinical trials, attention turns to strategies aimed at enhancing the likelihood of success at each stage. One of the strategies is druggability assessments which are done to improve the candidate’s drug-like qualities and raise the likelihood that their clinical development will be successful. Compounds that exhibit promise in this assessment are often chosen for further optimization, including medicinal chemistry modifications (1, 4).
Central to the pursuit of finding suitable drug candidates is the application of RO5 (5). The RO5 was proposed by Lipinski in 1997, which effectively guided the design of small molecule drugs over the subsequent 20 years (6). This rule has been widely used in medicinal research and a molecule that obeys the physicochemical property guidelines of the rule would be labelled as an ideal drug molecule (7). However, the growing diversity of drug compounds and therapeutic uses needs a more comprehensive strategy. Depending on the compound's pharmacological class and target profile, other essential descriptors such as bioavailability, solubility, and permeability may play an important role (8, 9).
Therefore, to widen the druggability assessment for all possible drugs using the RO5, a more practical approach is implemented by utilizing computational procedures that are based on virtual screening and ML. By leveraging computational techniques, such as ML to integrate the principles of RO5 into the drug screening processes, researchers can refine their selection of promising candidates with greater precision and efficiency (10). Despite the promise of computer approaches, druggability assessments remain challenging. Difficulties develop as a result of the requirement to balance numerous descriptors, such as molecular weight, lipophilicity, and hydrogen bonding properties, which differ greatly amongst drug compounds. Furthermore, these approaches can be data-intensive and require significant skill to provide accurate predictions, emphasizing the need for simple and effective ML-based solutions. These challenges emphasize the need for adaptive solutions that simplify the complexity of the assessment process while maintaining predictive accuracy (11, 12).
This study addresses these challenges by applying supervised ML algorithms, specifically Decision Tree (DT), Random Forest (RF), Linear Regression (LR), Naïve Bayes, k-Nearest Neighbor (k-NN), Support Vector Machine (SVM), and Extreme Gradient Boost (XGBoost). These algorithms are being applied to predict the druggability of pharmaceutical drugs. Previous research has explored druggability assessments using ML models (13, 14). However, studies frequently focus on a small number of descriptors or a single model type, which may not generalize well across different drug compounds. For example, some research focuses primarily on RO5 characteristics, while others use ML algorithms without completely integrating the druggability descriptors required for real-world applications across diverse pharmacological categories (13, 15, 16). The study’s originality stems from its approach to address these gaps by evaluating multiple ML algorithms alongside an expanded set of druggability descriptors. Through this approach, we aim to establish a more comprehensive and adaptable system for druggability prediction, allowing the assessment process to be both efficient and applicable to a wider range of drug compounds.
Literature Review
Introduction to Druggability Assessment
In the 1990s, the pharmaceutical industry became aware that clinical development was stopped because many of their compounds had unfavourable pharmacokinetics (PK) properties- essentially, how the body interacts with the administered substances throughout their presence (17-19). This realization prompted scientists to prioritize the optimization of lead compounds while considering these properties, thus needing techniques that could show the relationship between the PK properties and drug structures (18). Lead compounds that make it through the development process have been used to set the criteria for what causes the other compounds to fail during the drug development process. As a result, terms like ‘Druggable’ or ‘drug-like’ emerged to describe compounds deemed suitable for further development (20). However, assessing drug likeness alone is not comprehensive enough to measure a compound’s potential (20). Physicochemical characteristics such as molecular weight (MW), hydrophobicity, and polarity are found to preferentially occupy a relatively small range of potential values, according to the analysis of the observed distribution of these characteristics in authorized medications. Moreover, scrutiny extends to the relevance of drug targets, shedding light on disease mechanisms and facilitating the creation of precise therapeutic interventions (6, 21).
Failing to assess the druggability of a pharmaceutical compound can lead to consequences that are harmful in the process of drug development. One of the utmost significant effects is the wastage of valuable resources, including time and funding, on compounds that are unlikely to yield successful drugs (22). This misallocation of resources can hinder development in the field and delay the discovery of novel, effective medications. Compounds lacking suitable druggability characteristics might advance into clinical trials, where their shortcomings become apparent, resulting in substantial financial losses for pharmaceutical companies (23). Moreover, the risk of unexpected adverse effects in patients significantly increases when compounds with poor druggability interact unpredictably with biological systems (24).
Application and Limitations of Lipinski’s Rule
Preferential selection of compounds that are similar to drugs has been demonstrated to boost the probability of overcoming the high attrition rates in drug discovery (25). RO5 is most frequently utilized in practice in determining drug-likeness, aiding in the selection of compounds with a higher likelihood of success. This rule is a useful guideline in selecting drugs with good oral bioavailability and permeability and compounds that follow the criteria are more likely to be functionally sufficient to engage in significant interactions with proteins, which increases their potential as drug candidates (26). The rule specifies that, typically, an orally active drug should not have more than one violation of these criteria: a MW over 500 Da, a calculated logP (ClogP) over 5, more than five hydrogen bond donors, or more than ten hydrogen bond acceptors (nitrogen and oxygen atoms). Figure 1 illustrates the criteria of the Rule of Five. This rule, derived from the physicochemical parameters of 90% of orally active drugs that advanced to phase II clinical development, helps predict oral activity. RO5 is extensively utilized due to its simplicity and practicality. It offers a simple framework for assessing a compound's drug-likeness, making it simple to understand and utilize in drug discovery procedures (27-30).
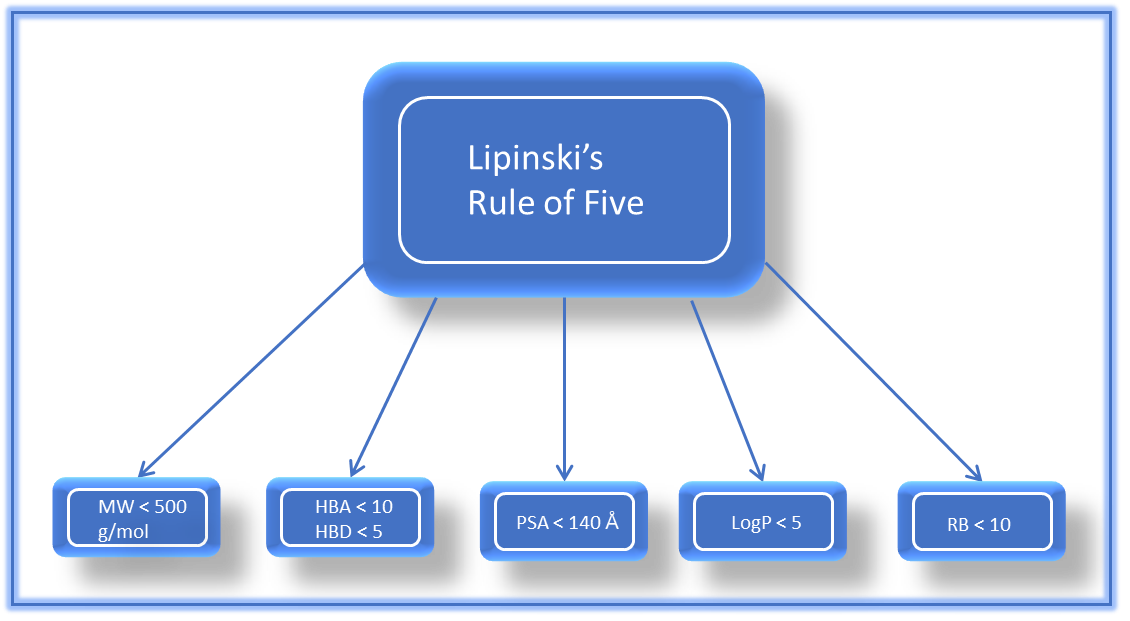
Despite its widespread use and ability to anticipate outcomes, the rule is not without limitations. A significant limitation is its exclusion of natural products and substrates for biological transporters from its criteria. Furthermore, according to the rule, compounds that violate more than one of these criteria are less likely to be orally active (27). However, the RO5 is crucial in assessing druggability for all drugs because it allows for preliminary screening of drug molecules that do not meet the criteria. It serves as an initial filter in small-molecule pharmaceutical screening, narrowing down the scope of drug candidates and reducing the costs associated with drug research and development (7). Thus, the RO5 guidelines have gained widespread adoption in the pharmaceutical industry as a rapid screening tool to identify compounds that have the potential to be developed into orally administered drugs (32). Therefore, to widen the druggability assessment for all possible pharmaceutical drugs using the RO5, researchers frequently employ computational tools such as virtual screening and ML in the early stages of drug discovery to assess these properties and prioritize compounds for further testing (33, 34).
Applications of Machine Learning in Drug Assessment
After exploring into the principles of drug assessment particularly through RO5, the focus now shifts towards innovative methodologies revolutionizing the field. These include ML, which represents a promising approach for discovering new drug molecules (35). Several ML algorithms and software have been developed and being utilized across all stages of drug discovery and development, comprising of clinical trials, identifying novel targets, improving the design and optimization of small-molecule compounds, developing new biomarkers, and increasing the understanding of disease mechanisms (36, 37).
In its most basic form, ML involves using algorithms to analyze data, learn from it, and make predictions or determinations about future data sets based on that learning. As the amount and caliber of data accessible for learning increases, the algorithms adaptively enhance their performance. ML is applied through two primary techniques: supervised learning and unsupervised learning. Unsupervised learning is used exploratively to construct models that cluster data without predefined categories, while supervised learning develop models to predict future values of categorical or continuous variables based on training data (38, 39).
Overview of Supervised Machine Learning Algorithms
Supervised ML, particularly, offers an important role in evaluating the pharmaceutical compounds’ druggability by examining input characteristics of chemical compounds, and predict crucial outcomes such as toxicity endpoints and biological activities (40). In this section, the overview of specific ML algorithms commonly employed in drug assessment will be explored, including DT, RF, NB, LR, k-NN, XGBoost, and SVM. A decision tree visually displays options and their outcomes in a tree-like structure (41). Each tree consists of the root, internal/test, and leaf nodes, each node represents classification attributes, and they also have branches that represent a value that the node can take (41, 42). The DT approach has been applied as a solution to the problems faced in designing combinatorial libraries, generating compounds for profiling, predicting biological activity, and predicting drug likeliness. DT is not only used for the identification of substructures that are given in the compound database to discriminate activity from non-activity, but it can be employed to classify chemical compounds as drugs or non-drugs (43).
A RF is a supervised ML algorithm built from DT algorithms. It is used for solving regression and classification problems by aggregating predictions from multiple decision trees. RF mitigate the shortcomings of individual decision trees, reducing overfitting and enhancing accuracy. They offer reliable predictions with minimal need for tuning parameters, making them highly valuable for drug assessment (40, 44).
LR is a regression technique in which the dependent variable is binomial or binary (45). LR is similar to Naïve Bayes in that it extracts weighted features from input data, takes logarithms of those features, and combines them linearly. Each feature is multiplied by a weight and then added together (42). The primary distinction between Naïve Bayes and LR lies in their classification approaches: Naïve Bayes is a generative classifier, while LR is discriminative. LR fits data to a logistic function to predict the probability of an event occurring. Like other forms of regression analysis, LR utilizes predictor variables that can be numerical or categorical (42).
Naïve Bayes is a classification approach that relies on Bayes’ theorem and assumes that all predictors are independent. This suggests that particular features present in a class are unrelated to other features present (41). Bayes’ theorem uses a mathematical framework (Equation 1) to explain the likelihood of an event that could have resulted from any two or more causes (43). The key focus of NB is the classification sector. Its main purpose is classification and clustering depending on the conditional probability of occurrence (41).
With P (A) = Probability of A occurring; P (B) = probability of B occurring; P (A/B) = probability of A given B; P (B/A) = probability of B given A.
Although the Bayesian concept has been around for a long time, its popularity as a tool in drug development and structure-activity research is relatively new (43). Naïve Bayes classifiers are used usually alongside or against other classifiers. Classifiers for Naïve Bayes are known for text filtering, but they are also employed in drug safety evaluation (40). It is mostly used in chemoinformatics for the prediction of biological properties as compared to physicochemical parameters. The practical application of these classifiers has been carried out for the prediction of toxicity in compounds, protein targets, and phospholipidosis mechanisms. It is also used in the classification of bioactivity for drug-like molecules (42).
The k-NN categorization approach allocates new compounds to the most prevalent class among known compounds in their vicinity. Proximity is determined by calculating Euclidean distances in a predefined feature space (45). k-NN can also perform regression and is considered one of the fundamental machine-learning algorithms. SVM is another algorithm highlighted for its utility with noisy data (45). It is a complex algorithm, but it prevents theoretical guarantees concerning data overfitting and can provide high accuracy (46). XGBoost is an ML technique adept at handling regression and classification tasks. It iteratively builds a set of weak learners to produce a strong predictive model, showcasing its effectiveness in drug assessment (47).
Related Work and Research Gaps
Studies have utilized various models to predict molecular properties, such as the study by Ståhl and colleagues, where they introduced a flexible deep Convolutional Neural Network (CNN) method. This approach is designed for analysing graph structures of arbitrary sizes that represent molecules (46). Wen and co-workers proposed deep learning-based models for predicting molecular properties by extracting features from various representations of molecules (48). While Meyer and colleagues used RF model and CNN models to evaluate drug classification methods based on chemical structure-derived images (35). Hannamm et al. , evaluated drug reactions using DT modelling, calculating a Tanimoto coefficient of 0.85 to demonstrate the significant structural diversity within their dataset (49). Shi et al. , utilized RF to predict drug-target interactions, assessing model performance through 5-fold cross-validation (50). Sugaya and Ikeda assessed the druggability of protein-protein interaction and predicted novel druggable using SVM (51).
Numerous challenges arise when attempting to utilize RO5 within ML frameworks to predict the druggability of pharmaceutical compounds. While existing research employs diverse ML models such as deep CNN, DT, RF, SVM, and Lasso combined with RF, there remains a significant absence of a comprehensive comparative analysis across these models, specifically concerning the prediction of druggability based on RO5. Therefore, the goal of this research is to undertake a comprehensive comparison of various ML algorithms. The analysis will primarily focus on assessing the efficacy of applying RO5 for druggability prediction, while also pinpointing the strengths and weaknesses inherent in each model.
Theoretical Framework
The theoretical framework presented integrates ML techniques into the context of druggability assessment in drug discovery, with a particular emphasis on RO5. This framework provides background information on druggability assessment, RO5, and ML algorithms (52).
| Molecular Descriptor | Description | Software used for calculation |
|---|---|---|
| Molecular Weight | Molecular Weight | RDKit |
| HBD | Number of Hydrogen Donors | RDKit |
| HBA | Number of Hydrogen Acceptors | RDKit |
| LogP | Log of 1-octanol/water partition co-efficient (neutral form) | RDKit |
| TPSA | Total Polar Surface Area | RDKit |
| Num_Rotatable_Bonds | Number of Rotatable Bonds | RDKit |
| SAS | Synthetic Accessibility Score | RDKit |
Methodology
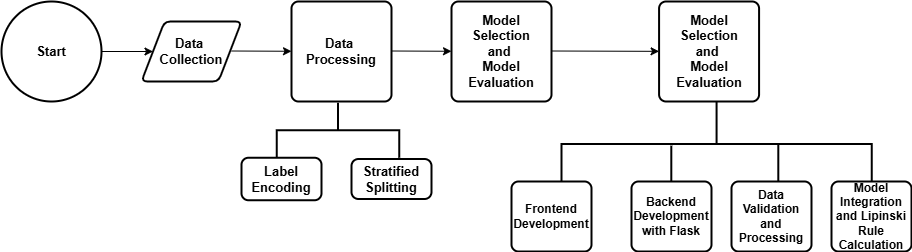
This research assessed the druggability of pharmaceutical compounds employing RO5 with ML models. The methodology was carried out following the process of data collection, data processing, model selection, model evaluation, and subsequent application development which is summarised in Figure 2. The methods are discussed below in detail to offer insights into the predictive capacity of ML algorithms in determining pharmaceutical compound druggability, thereby contributing to the advancement of drug research and development methodologies.
Data Collection
In this study, the data was collected by downloading it directly from the DrugBank site (https://go. drugbank. com/releases/latest#structures). The data contained structure information represented as InCHI, InCHI Key, and SMILES notation. The compounds’ SMILES notation was utilized with the Lipinski RDKit module in Python, an open-source cheminformatics software that facilitates the integration of comprehensive molecular information. All the properties used to determine if a molecule passes the Lipinski Rules were calculated/extracted using this module and these included molecular descriptors as illustrated in Table 1 below.
Data Processing
Subsequent to the collection of the data, columns with missing and irrelevant data were dropped, resulting in a dataset of 11 583 columns. Libraries like RDKit were used to compute molecular descriptors such as HBD, HBA, TPSA, MW, LogP, SAS, and Num_Rotatable_Bonds. These descriptors were employed to evaluate if pharmaceutical compounds adhere to Lipinski’s Rule, represented by the binary target variable "PassesLipinski". Label encoding was applied to convert "PassesLipinski" into numerical values: 1 for true (adheres to the RO5) and 0 for false (violates the RO5).
Label Encoding
For the binary target variable, ‘PassesLipinski’, label encoding was used to represent the classes as 0 and 1, where 1 indicates that a drug compound adheres to RO5, and 0 indicates non-adherence. This transformation allows ML models to process the target variable effectively. Enabling classification algorithms to learn patterns associated with Lipinski Properties.
Stratified Splitting
The dataset was split into training and testing sets using the ‘train_test_split’ function from scikit-learn, with 80% of the molecules allocated for training the model and 20% for testing (Table 2). Stratified splitting was employed to maintain the class distribution of the target variable ‘PassesLipinski’ in both sets. This method is particularly crucial for handling imbalanced datasets, where one class (in this case, compounds passing Lipinski Properties) might be significantly smaller than the other class. By maintaining the proportion of classes in both sets, stratified splitting helps prevent biased model performance evaluation. It ensures that the models are trained and tested on representative samples from both classes, leading to more reliable assessments of their predictive capabilities.
| Total Compounds | TrainingSet (80%) | TestingSet (20%) |
|---|---|---|
| 11 583 | 9253 | 2314 |
These preprocessing steps were essential to preparing the data for model training and evaluation. Label encoding facilitated the numerical representation of categorical variables, while stratified splitting enhanced the robustness of the models by preserving the inherent class distribution within the dataset.
Model Selection and Model Evaluation
Classification was the main approach in this study and different algorithms were developed to evaluate which model would be suited for the classification of drugs to predict whether they are druggable or not. These models consisted of DT, LR, RF, k-NN, XGBoost, SVM, and Naïve Bayes. These models were trained and tested utilizing identical parameters.
Relevant measures namely balanced accuracy, precision, F1-score, recall, and Receiver Operating Characteristics-Area Under the Curve (ROC-AUC) score were utilized in evaluating the performance of the developed models. The models were fine-tuned by adjusting hyperparameters. The hyperparameter grid was defined and a grid search object was created. The evaluation metric was defined, and accuracy was a chosen performance metric for optimization.
The Development of DrugCheck Master Application
Frontend Development
In the frontend development, HTML, CSS, and JavaScript were combined to create visually appealing and user-friendly interfaces. HTML structured web page elements, CSS enhanced visual appeal, and JavaScript added dynamic interactivity. Input fields, crucial for user interaction, were implemented using JavaScript within HTML forms. JavaScript ensured input validation, maintaining data integrity by enforcing specific criteria like syntax and allowable characters.
Backend Development with Flask
In the backend development, Flask, a Python web framework, managed server-side logic and defined API endpoints. Flask applications handled diverse backend operations by defining routes for various URL endpoints. For example, (/check_lipinski) route processed POST requests containing compound data from the frontend securely. Flask's handling of requests ensured accurate and secure data transmission. Within routes, data extraction logic parsed incoming data for effective processing. Flask facilitated seamless integration with external APIs or databases, enriching analysis with detailed compound information based on user input. This enhanced the application’s predictive capabilities and assessment accuracy.
Data Validation and Processing
In Flask, meticulous data validation ensured valid compound names or formulas, preserving data integrity. Integration with external APIs provided detailed compound information for analysis, enriching assessments. Chemical features like MW, logP, TPSA, and hydrogen bond counts were systematically extracted to evaluate adherence to RO5.
Model Integration and Lipinski Rule Calculation
The pre-trained RF ML model was seamlessly integrated into the Flask application, converging data analysis and computational modeling for precise predictions. Trained on diverse datasets, the model comprehended complex chemical features like MW, logP, TPSA, and hydrogen bond counts. These features provided complex insights into pharmaceutical compounds’ physicochemical properties, crucial for evaluating adherence to RO5. The model applied sophisticated algorithms to assess Lipinski rule compliance, predicting druggability outcomes as either “pass” or “fail”.
Result and Discussion
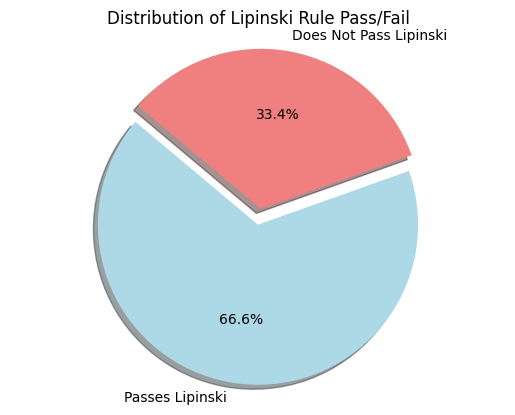
A comparison of diverse supervised ML algorithms is recommended for performance review. Various ML models may result in different outcomes which indicate better or worse performance for each model. In this study, seven models- RF, DT, K-NN, XGBoost, Naïve Bayes, LR, and SVM classifier were used to classify if chemical compounds pass or fail Lipinksi’s rules. Before evaluating the performance of the models, visualization of the features was done where some of the structures were extracted using SMILES strings, and the information on the features was extracted. Figure 3 below shows examples of some of the chemical compounds present in the evaluated database. Figure 4 illustrates compounds categorized by their adherence to RO5: 33.4% of the chemical compounds failed RO5 meaning that they violated all the Lipinski’s properties. While approximately 66.6% of the compounds passed RO5 meaning the compounds comply with two or more properties (18). These percentages were drawn from all the compounds in the evaluated database. This means that the analysed database contained both fractions of compounds that adhere to and violate RO5.
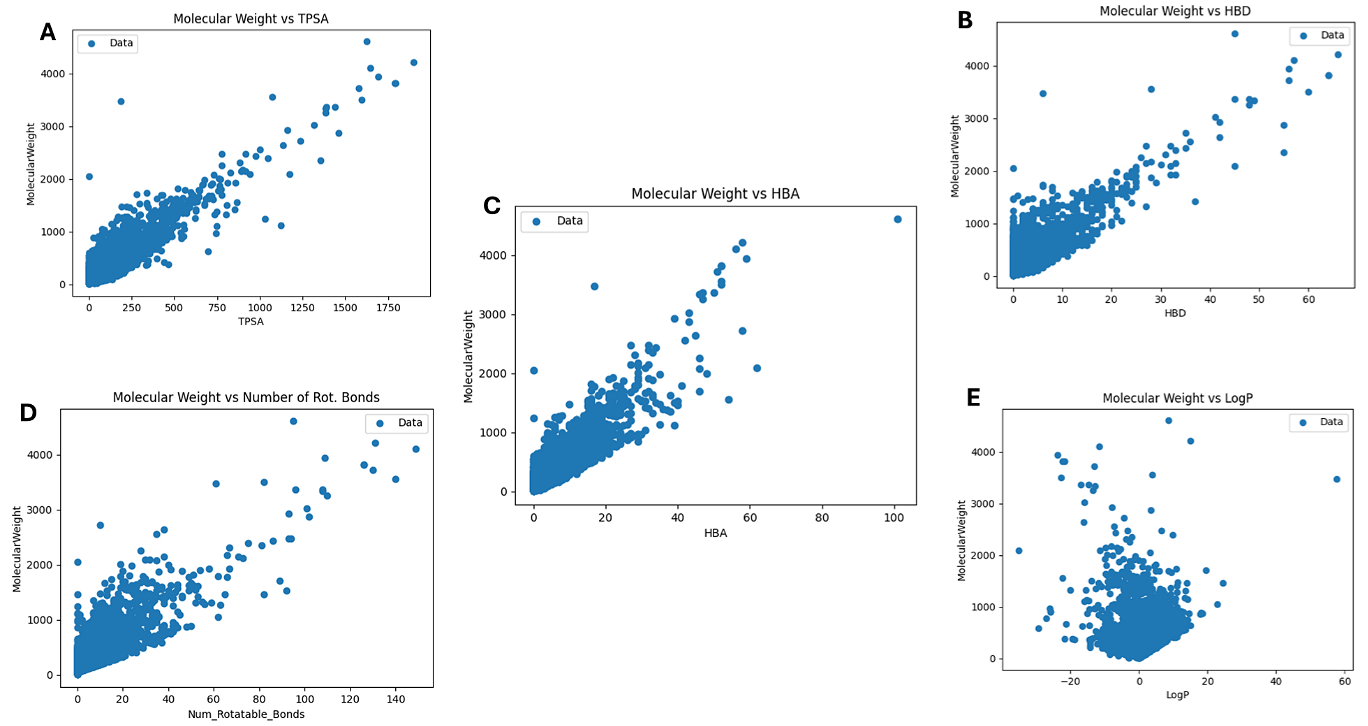
To visualize the relationship between Lipinski's properties, plots were created, and the results are displayed in Figure 5. All the attributes were plotted against the MW. By plotting the other RO5s against the MW, it allows for the observation of potential trends on how drug-likeness and bioavailability are affected by increasing molecular sizes. Molecules with higher MW typically have more complex structures, which may have an impact on their PK properties.
The analysis of HBA, HBD, LogP, and rotatable bonds change with MW can reveal correlations or patterns. For example, larger molecules may include more rotatable bonds, higher TPSA and a higher number of donors and acceptors, leading to higher lipophilicity (higher LogP). This was observed in the simulated figures. Figure 5 (A-E) illustrate that as MW increases, so do the corresponding features and the analysis of the data indicated that compounds weighing less than 1000 Da generally had between 0 and 10 HBDs and a similar pattern was observed for HBAs. A study by Rashid et al. , (2021), revealed that TPSA predicts drug transport parameters including adsorption and brain penetration, and that compounds with TPSA < 140 Ų are generally believed to have good cell membrane permeability where higher TPSA values may be indicative of better solubility but potentially poor permeability (53, 54). In this study, most molecules with a MW of less than 1000 Da exhibited a TPSA value between 0 and 250, indicating potential interactions with biological membranes and proteins (54). For the LogP values, which ranged from -20 to 20 in molecules with MW less than 1000 Da, indicated a vast range of hydrophilicity to lipophilicity. Atkinson et al. , (2021), revealed that extremely low logP values (negative values) suggest high hydrophilicity, potentially resulting in poor membrane permeability and very high logP values (positive values) suggest high lipophilicity, which may result in poor solubility in aqueous environments and potential bioavailability issues (55). The results found in this study show that a significant number of the analyzed compounds likely comply with or are close to Lipinski’s guidelines. The number of rotatable bonds ranged between 0 and 40 for molecules with MW less than 2000 Da, highlighting a wide variety of molecular flexibilities. Generally, compounds with fewer rotatable bonds (< 10) are preferred for drug development due to better bioavailability and more efficient binding to biological targets (56). Visualizing these properties in Figures 3 – 5, helps identify promising candidates and guides optimization efforts in drug development to enhance drug-likeness.
Model Performance Evaluation
The results presented in this study were obtained through a contrast of several ML binary classifiers and different performance metrics were evaluated. These included balanced accuracy, precision, recall, F1-Score, and Area under the Receiver Operating Characteristic Curve (ROC-AUC) score. The main accuracy metric used was the balanced accuracy obtained on the test data. Recall, precision, and F1-Score were additionally computed for the different models. The classification quality was then assessed for each model using the AUC plot. The AUC is a measure that indicates how good a classifier is at performing a specific classification task. The AUC value ranges between 0 and 1, with an efficient classifier having an AUC value near 1. Table 3 details the performance metric values obtained in this study for the different supervised ML models evaluated.
| Model | Balanced accuracy | Precision | Recall | F1-Score | ROC-AUC Score |
|---|---|---|---|---|---|
| DT | 99.87% | 99.87% | 1.0 | 99.93% | 99.87% |
| RF | 99.94% | 99.81% | 1.0 | 99.90% | 99.94% |
| KNN | 93.28% | 94.59% | 0.98 | 96.10% | 93.28% |
| SVM | 82.63% | 87.76% | 0.90 | 89.02% | 82.63% |
| XGBoost | 99.81% | 99.81% | 1.0 | 99.90% | 99.81% |
| LR | 83.70% | 87.70% | 0.94 | 90.51% | 83.70% |
| NB | 81.94% | 84.77% | 0.99 | 91.51% | 81.94% |
The evaluation highlighted the prowess of various trained models, notably the DT, SVM, RF, XGBoost, and logistic regression. Among these, the RF, DT, and XGBoost exhibited exceptional performance, boasting an impressive balanced accuracy of > 99%. Notably, this accuracy threshold was crucial, leading to a focused analysis on models surpassing the 95% accuracy mark. RF showed the highest accuracy of 99.94%, while the XGBoost model showed excellent results with a precision of 99.81%. The Recall showed good results for DT, RF, LR, and XGBoost. These models are particularly noteworthy for their high recall, indicating their sensitivity and reliability in identifying true positives. Conversely, LR, SVM, and NB showed accuracies below 95% with NB having the least accuracy of 81.94%. The results suggest that for applications requiring high accuracy, balanced accuracy, recall, and precision, RF, DT, and XGBoost are the most suitable models.
The performance of the models with high sensitivity and reliability (DT, RF, and XGBoost) was further evaluated using the Receiver Operating Characteristics, which is a graphical representation employed to evaluate the classification model’s performance. Figure 6 present the Receiver Operating Characteristics for the RF, DT, and XGBoost models respectively. The curves show the AUC-Score calculated for each model.
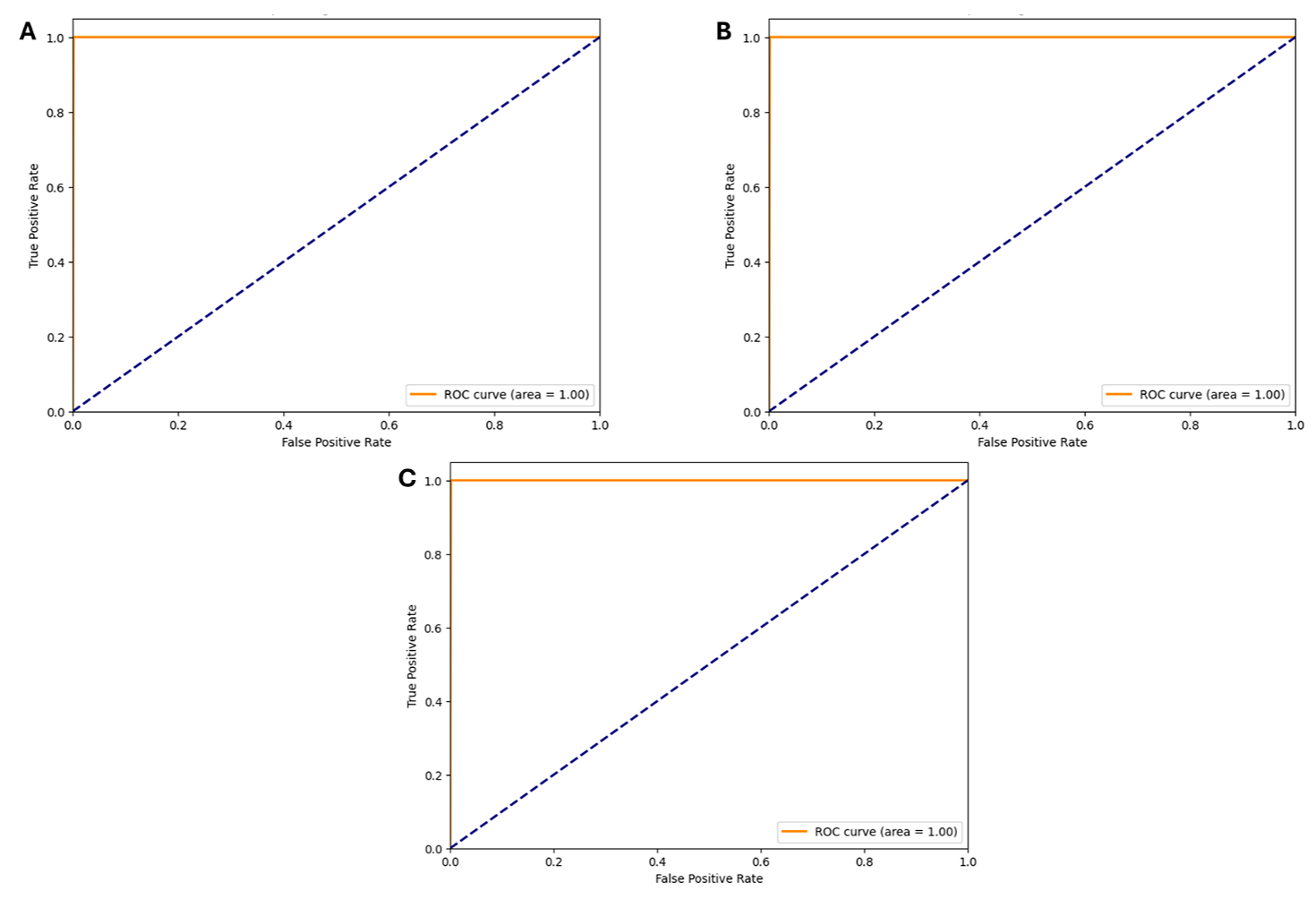
The DT, RF, and XGBoost models achieved an AUC of 1.00 and a ROC curve of 1.00 is a perfect ROC curve, indicating that the model has achieved perfect discrimination between the positive and negative classes, illuminating their efficacy in discerning Lipinski rule compliance. Liu et al. , (2020), found that models that achieve a higher accuracy and ROC-AUC score have a high predictive performance. Within this study, these ensemble models with better scores outperformed the other models, demonstrating their adaptability in dealing with complex, high-dimensional chemical data as well as their capacity to reveal precise correlations between molecular properties and desired outcomes (57-59).
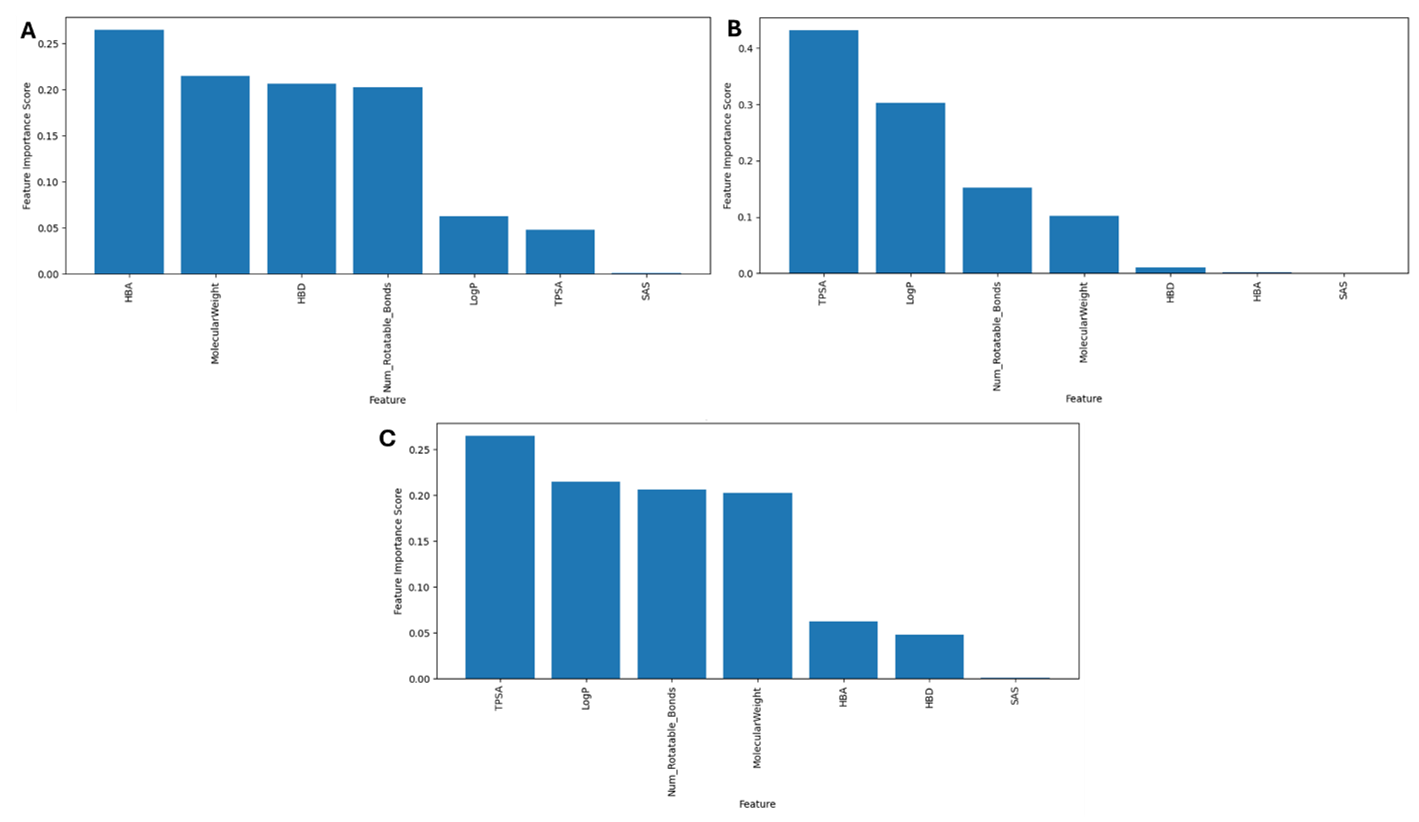
The goal of feature selection is to prevent overfitting, enhance model performance, and gain a better understanding of the underlying data generation processes. RF inherently conducts feature selection as it constructs classification rules (60). Feature importance is utilized to rank the importance of features within a dataset, and for the RF model, the HBA feature was the most important feature showing its significance in distinguishing druggable from non-druggable compounds. Following HBA, the MW, HBD, and number of rotatable bonds also displayed significant importance. These findings indicate that molecular properties such as hydrogen bonding capacity, size, and flexibility play crucial roles in determining drug druggability. The SAS feature had the lowest score, indicating that it had less of an impact on the model’s ability to predict (Figure 7A).
In the DT model, the feature property having the highest score is the HBD feature. This indicates that the number of hydrogen atoms available for forming bonds could display an important role in determining a drug’s druggability (13). This is followed by MW, HBA, and number of rotatable bonds. While TPSA, LogP had the lowest scores indicating they might not be as influential in this model. The absence of an importance score for the SAS feature indicates that it may not have a significant influence on the predictive performance of the model (Figure 7B).
The TPSA feature had the highest score for the XGBoost model, suggesting its significance in distinguishing between druggable and non-druggable drugs. This shows that the feature is important in determining the drug’s pharmacokinetic and pharmacodynamic properties, which influence their efficacy and safety (13). Following TPSA, the LogP, MW and number of rotatable bonds also displayed significant importance. Features like HBD, HBA were the lowest suggesting that while these properties may still contribute to druggability prediction, they might not be as influential as other factors while SAS did not have an importance score (Figure 7C). Amongst the models, TPSA, LogP, number of rotatable bonds and molecular weight frequently appeared as top predictors and this can be due to their direct influence on the PK properties and their effects on a compound’s ability to be absorbed, distributed, metabolize, and excreted (56, 61).
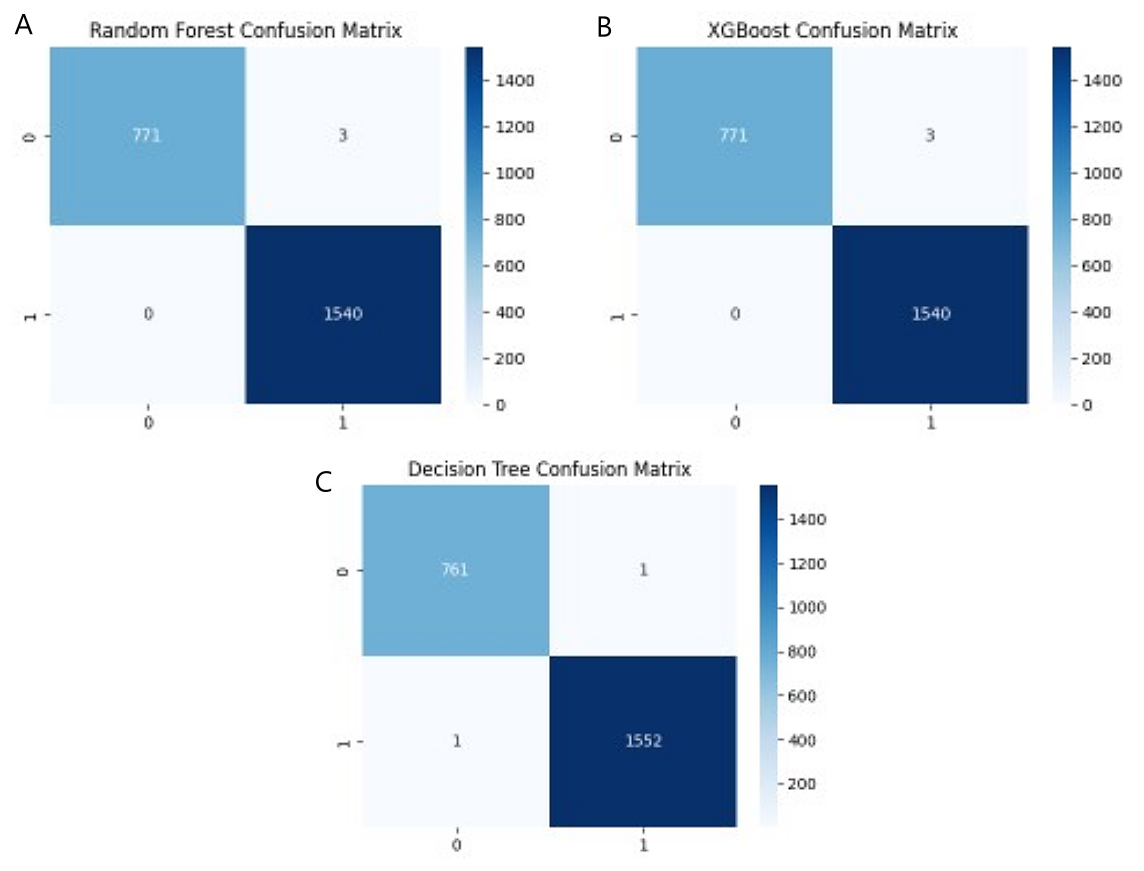
The inclusion of the confusion matrix diagrams provides additional insights into their performance, facilitating a comprehensive evaluation of the model’s predictive capabilities (Figure 8). The RF and XGBoost models had a total of 771 true positives, 3 false positives, 0 false negatives, and 1 540 true negatives. These findings highlight the excellent accuracy and robustness of both ensemble methods, demonstrating their ability to effectively reduce misclassifications. In comparison, the DT model exhibited a significantly lower true positive count of 761 and true negative count of 1 552, with one false negative and one false positive, demonstrating slightly lower predictive precision and recall than the ensemble models.
Research has shown that Random Forest (RF) and XGBoost outperform other models in handling complex feature interactions and imbalanced data (62). Our results confirm this trend, with both RF and XGBoost achieving a zero false-negative rate, consistently identifying positive cases—an essential feature in druggability prediction. Model choice depends on factors like computational efficiency, interpretability, and the application’s needs. For example, RF is preferred when model transparency is critical, while XGBoost and Decision Trees (DT) may be better for complex datasets requiring fine-tuning. RF also provides a balance between accuracy and interpretability, making it highly adaptable. XGBoost’s precision and customization options are beneficial for refining predictions in high-dimensional data.
| Parameters | Goselerin | Phenylalanine | Aspirin |
|---|---|---|---|
| Properties | HBD: 17HBA: 16MW: 1269.433LogP: -3.1057TPSA: 495.89 | HBD: 2HBA: 2MW: 165.192LogP: 0.641TPSA: 63.32 | HBD: 1HBA: 3MW: 180.16LogP: 1.4TPSA: 140 |
| Result | False | True | True |
| Observation | Goserelin's high number of hydrogen bond donors (HBD=17) and hydrogen bond acceptors (HBA=16) exceeds Lipinski's recommended limits. Its large molecular weight (MW=1269.433) also surpasses the rule's threshold. Furthermore, its negative LogP value (-3.1057) indicates high hydrophilicity, which is contrary to Lipinski's guideline (LogP ≤ 5). Based on these properties, Goserelin does not adhere to Lipinski's rule, as indicated by the "false" result. Violation of these criteria might suggest challenges related to its oral bioavailability and could impact its druggability. | Phenylalanine has properties well within Lipinski's guidelines. It has a low number of hydrogen bond donors (HBD=2) and a moderate number of hydrogen bond acceptors (HBA=2). Its molecular weight (MW=165.192) and LogP value (0.641) fall within the acceptable ranges. Consequently, Phenylalanine adheres to Lipinski's rule, indicated by the result "true". Its compliance with Lipinski's criteria suggests that Phenylalanine has favourable properties for oral bioavailability and potential drug development. | Aspirin, has properties within Lipinski's guidelines. It has a low number of hydrogen bond donors (HBD=1) and a moderate number of hydrogen bond acceptors (HBA=3). Its molecular weight (MW=180.16) and LogP value (1.4) fall within the acceptable ranges. |
The RF model, following an extensive period of rigorous training and optimization, demonstrated a remarkable accuracy rate of 99.9% in accurately categorizing compounds as either compliant or non-compliant with RO5. This achievement was further bolstered by the ROC curve, as illustrated in Figure 9, where an AUC of 1.00 was observed. The ROC curve's proximity to the top-left corner of the plot suggests an exceptional performance of the model. The indicated positioning signifies that the model attains a high true positive rate (sensitivity) while simultaneously maintaining a low false positive rate. Such characteristics underscore the RF model's robustness in distinguishing between compounds that adhere to RO5 and those that do not, making it a highly reliable tool in the prediction of druggability. A comparative study by Sagi and Rokach, (2018) found that RF and XGBoost displayed better balanced accuracy with 99% compared to other models such as DTs, and SVM (63). In a specific application to drug discovery, Chen and Guestrin (2016) demonstrated that XGBoost and RF consistently achieved the highest accuracy among various datasets, surpassing SVM, DT, and LR (64). This analysis supports earlier findings that RF and XGBoost outperform all other models in terms of accuracy.
DrugCheck Master Application
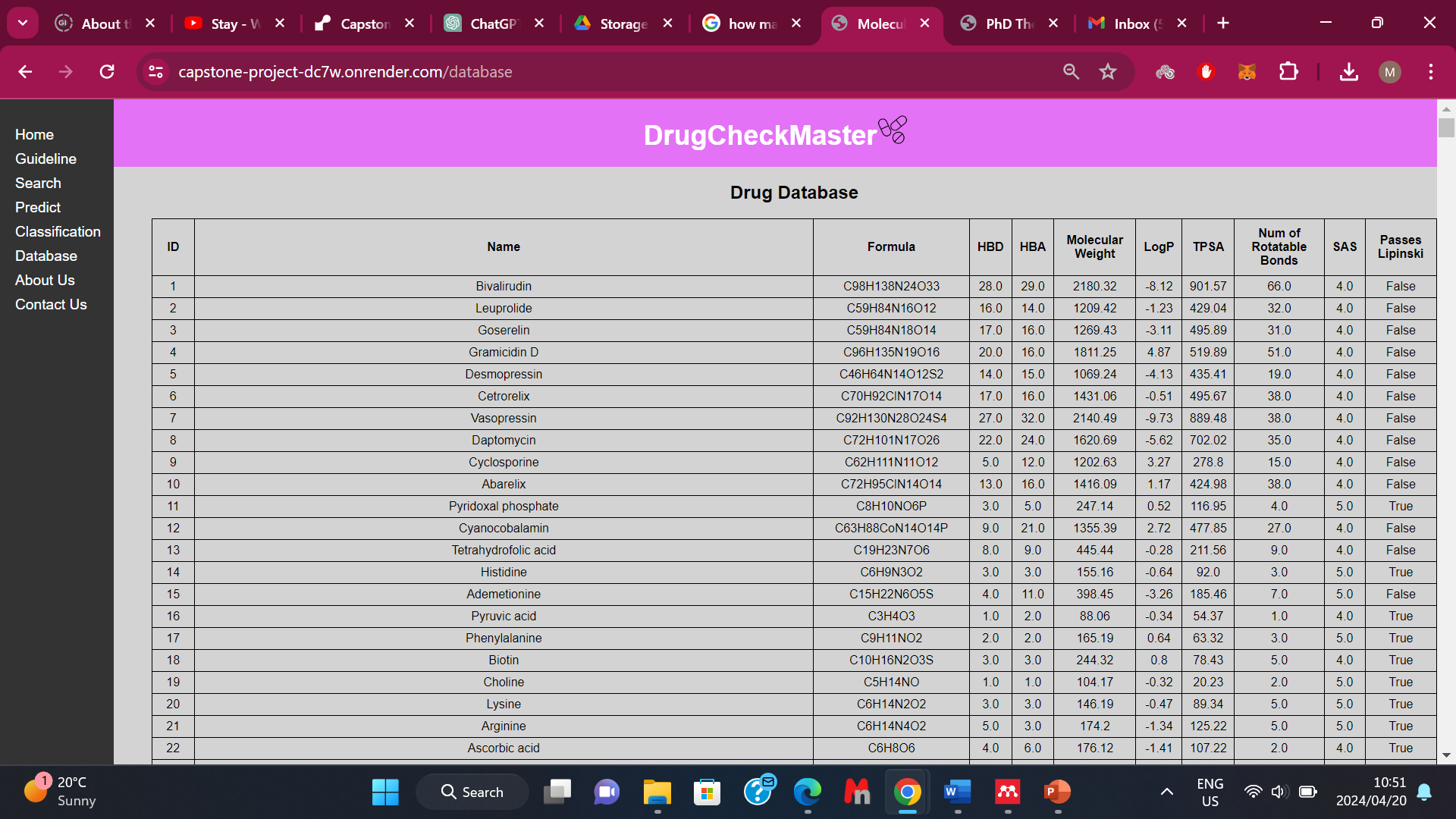
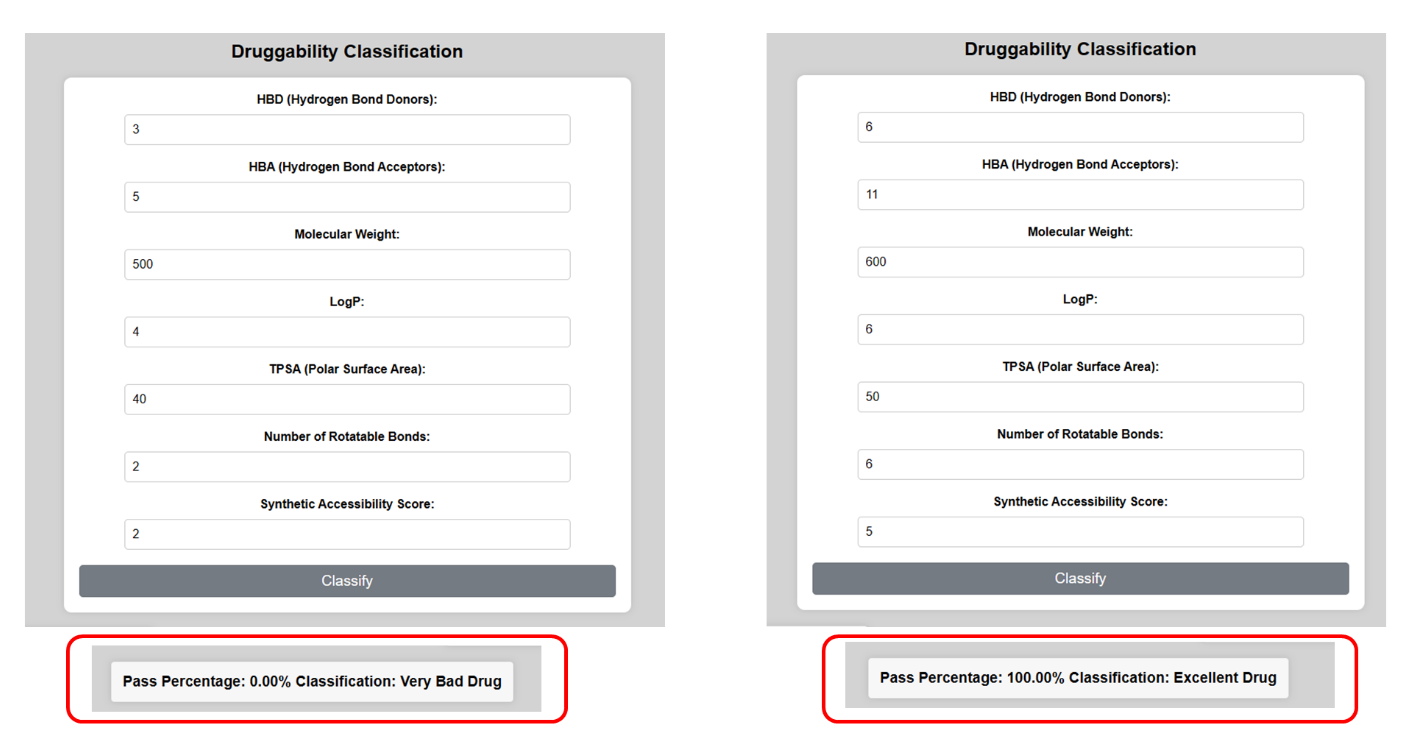
Using the RF model which had a highly balanced accuracy and the ability to predict both classes effectively, an application was built to predict the Lipinski Properties and also classify if a molecule is druggable or not. It also has a database section where it contains the chemical compound’s information such as the formula, and all the Lipinski’s physicochemical properties (Figure 9). This application was deployed on Render which is a cloud application platform and can be accessed via the link provided in the Data Availability section.
Rule of Five for some compounds present in our database was observed and interesting outcomes based on their properties were shown in the application (see Figure 10). Some of the drugs assessed for the rule using the application built using RF as a model include Goserelin, Aspirin, and Phenylalanine, the details obtained using the application are detailed in Table 4.
In the evaluation of the pharmaceutical compounds from Table 4, Goserelin and Phenylalanine using RO5, intriguing outcomes were observed based on their specific properties. Goserelin, with a MW well below the threshold at 384, satisfies the first condition of RO5. Its partition coefficient (LogP) of -0.83 falls significantly below the permissible limit of 5, indicating excellent hydrophilicity. Moreover, Goserelin exhibits a low count of hydrogen bond donors (HBD = 10) and acceptors (HBA = 19), surpassing stipulated maximum values. However, its topological polar surface area (TPSA) at 338.22 Ų exceeds the allowable limit of 140 Ų. This discrepancy implies that while Goserelin adheres to several of Lipinski's criteria, its TPSA value might pose challenges to its oral bioavailability. Brown (2020) revealed that Goserelin is a synthetic decapeptide that mimics luteinizing hormone-releasing hormone (LHRH) which is mostly used in cancer treatment particularly prostate and breast cancer. The study further discussed that the druggability of Goserelin is high due to its specific mechanism of action despite the fact that it adheres few Lipinski rule, drugs like this are often injected rather than orally to avoid degradation in the gastrointestinal tract (65).
Conversely, Phenylalanine, a common amino acid with a MW of 165.192 g/mol, exhibits properties that align more closely with RO5. It comfortably meets the MW requirement, its LogP value of 1.89 suggests moderate lipophilicity, well within the permissible range. Additionally, Phenylalanine showcases minimal donors (HBD = 2) and acceptors (HBA = 3), indicating favourable properties for druggability. Notably, its TPSA of 37.3 Ų underscores its relatively compact molecular structure, further supporting its potential as a viable drug candidate. However, Phenylalanine is not typically considered a drug on its own (65). The study by Ciobanu et al. , (2023) agrees with previous studies and further revealed that Phenylalanine role in drug formulations is more about its metabolic importance and incorporation into larger, biologically active molecules and its obedience with RO5 suggests good oral bioavailability when used as part of a drug formulation/molecule (66).
Table 4 shows that Phenylalanine and aspirin align with Lipinski's guidelines and are more likely to be a viable drug candidate, Goserelin's properties violate these rules, highlighting potential challenges in its drug development process. Adherence to RO5 aids as an effective initial screening criterion during the selection and prioritization of compounds for further pharmaceutical development. Goserelin does not adhere to RO5 indicated by the result - "false". The violation of these criteria might suggest challenges related to its oral bioavailability and could impact its druggability.
Conclusion
In conclusion, this study displayed the efficacy of ensemble learning techniques, such as RF, XGBoost, and Decision Tree in evaluating pharmaceutical compounds' adherence to Lipinski's Rule. These models achieved near-perfect accuracy, providing reliable identification of drug-like properties. The DrugCheckMaster application was developed utilizing the most predictive ML model, which allows for more efficient and scalable drug candidate evaluation, facilitating early-stage drug discovery. By harnessing ML, pharmaceutical researchers can streamline drug discovery processes, leading to more targeted and efficient development of therapeutics. To improve accuracy, integrating additional physicochemical properties and utilizing model combination strategies is recommended to mitigate potential underfitting or overfitting. These approaches have proven effective in improving the classification of drugs into druggable and non-druggable groups.
Declarations
Conflict of Interest
The authors declare no conflicting interest.
Data Availability
The unpublished data is available upon request to the corresponding author. The application is freely available at the link: https://capstone-project-dc7w.onrender.com/
Ethics Statement
Not applicable.
Funding Information
Not applicable.
References
- Sinha S, Vohora D. Drug Discovery and Development: An Overview. In: Pharmaceutical Medicine and Translational Clinical Research. Elsevier Inc. ; 2017. p. 19– 32.
- Grow C, Gao K, Nguyen DD, Wei GW. Generative network complex (GNC) for drug discovery. Commun Inf Syst. 2019; 19 (3): 241– 77.
- Nicolaou KC. Advancing the Drug Discovery and Development Process. Angewandte Chemie. 2014 Aug 25; 126 (35): 9280– 92.
- Yuan JH, Han SB, Richter S, Wade RC, Kokh DB. Druggability Assessment in TRAPP using Machine Learning Approaches. J Chem Inf Model [Internet]. 2020; 60 (3): 1685– 99. Available from: https://doi. org/10.1101/2019.12.19.882340
- Staszak M, Staszak K, Wieszczycka K, Bajek A, Roszkowski K, Tylkowski B. Machine learning in drug design: Use of artificial intelligence to explore the chemical structure– biological activity relationship. Vol. 12, Wiley Interdisciplinary Reviews: Computational Molecular Science. John Wiley and Sons Inc; 2022.
- Wei W, Cherukupalli S, Jing L, Liu X, Zhan P. Fsp3: A new parameter for drug-likeness. Vol. 25, Drug Discovery Today. Elsevier Ltd; 2020. p. 1839– 45.
- Chen X, Li H, Tian L, Li Q, Luo J, Zhang Y. Analysis of the Physicochemical Properties of Acaricides Based on Lipinski’ s Rule of Five. Journal of Computational Biology. 2020 Sep 1; 27 (9): 1397– 406.
- Price E, Weinheimer M, Rivkin A, Jenkins G, Nijsen M, Cox PB, et al. Beyond Rule of Five and PROTACs in Modern Drug Discovery: Polarity Reducers, Chameleonicity, and the Evolving Physicochemical Landscape. J Med Chem. 2024 Apr 11; 67 (7): 5683– 98.
- Li B, Wang Z, Liu Z, Tao Y, Sha C, He M, et al. DrugMetric: quantitative drug-likeness scoring based on chemical space distance. Brief Bioinform. 2024 Jul 1; 25 (4).
- Castello FA. Preselection of Compounds for Lead Identification in Virtual Screening Campaigns. In: Marti MA, Turjanski AG, Do Porto DF, editors. Structure-based drug design2. Springer; 2024. p. 109– 25.
- Niazi SK, Mariam Z. Computer-Aided Drug Design and Drug Discovery: A Prospective Analysis. Pharmaceuticals. 2024 Jan 1; 17 (1).
- Chang Y, Hawkins BA, Du JJ, Groundwater PW, Hibbs DE, Lai F. A Guide to In Silico Drug Design. Vol. 15, Pharmaceutics. MDPI; 2023.
- Agoni C, Olotu FA, Ramharack P, Soliman ME. Druggability and drug-likeness concepts in drug design: are biomodelling and predictive tools having their say? Vol. 26, Journal of Molecular Modeling. Springer; 2020.
- Jamali AA, Ferdousi R, Razzaghi S, Li J, Safdari R, Ebrahimie E. DrugMiner: Comparative analysis of machine learning algorithms for prediction of potential druggable proteins. Vol. 21, Drug Discovery Today. Elsevier Ltd; 2016. p. 718– 24.
- Imani A, Agung Santoso Gunawan A, Suhartono D. Interpretable Machine Learning in Drug Discovery: QSAR Modeling of Molecular Properties for Alzheimer’ s Disease Using Random Forest. International Journal of Computing and Digital Systems [Internet]. 2024 May 10; 1– 10. Available from: http://journals. uob. edu. bh
- Chithrananda S, Grand G, Ramsundar B. ChemBERTa: Large-Scale Self-Supervised Pretraining for Molecular Property Prediction. 2020; 1– 7. Available from: https://ml4molecules. github. io
- M. Honorio K, L. Moda T, D. Andricopulo A. Pharmacokinetic Properties and In Silico ADME Modeling in Drug Discovery. Med Chem (Los Angeles). 2013 Jan 1; 9 (2): 163– 76.
- Bickerton GR, Paolini G V. , Besnard J, Muresan S, Hopkins AL. Quantifying the chemical beauty of drugs. Nat Chem. 2012 Feb; 4 (2): 90– 8.
- Grogan S, Preuss C V. Pharmacokinetics. 2024.
- Lobo S. Is there enough focus on lipophilicity in drug discovery? Vol. 15, Expert Opinion on Drug Discovery. Taylor and Francis Ltd; 2020. p. 261– 3.
- Chen X, Li H, Tian L, Li Q, Luo J, Zhang Y. Analysis of the Physicochemical Properties of Acaricides Based on Lipinski’ s Rule of Five. Journal of Computational Biology. 2020 Sep 1; 27 (9): 1397– 406.
- Parvathaneni M, Awol AK, Kumari M, Lan K, Lingam M. Application of Artificial Intelligence and Machine Learning in Drug Discovery and Development. Journal of Drug Delivery and Therapeutics. 2023 Jan 15; 13 (1): 151– 8.
- Raghavendra NM, Kumar BP, Sasmal P, Teli G, Pal R, Gurubasavaraja Swamy PM, &, et al. Designing Studies in Pharmaceutical and Medicinal Chemistry. In The Quintessence of Basic and Clinical Research and Scientific Publishing. In: The Quintessence of Basic and Clinical Research and Scientific Publishing. 2023. p. 125– 48.
- Ritchie TJ, Macdonald SJF. How drug-like are ‘ ugly’ drugs: do drug-likeness metrics predict ADME behaviour in humans? Drug Discov Today. 2014 Apr; 19 (4): 489– 95.
- Hu Q, Feng M, Lai L, Pei J. Prediction of Drug-Likeness Using Deep Autoencoder Neural Networks. Front Genet. 2018 Nov 27; 9.
- Ntie-Kang F, Nyongbela KD, Ayimele GA, Shekfeh S. “ Drug-likeness” properties of natural compounds. Physical Sciences Reviews. 2019 Nov 26; 4 (11).
- Protti Í F, Rodrigues DR, Fonseca SK, Alves RJ, de Oliveira RB, Maltarollo VG. Do Drug-likeness Rules Apply to Oral Prodrugs? ChemMedChem. 2021 May 6; 16 (9): 1446– 56.
- Ahmad I, Kuznetsov AE, Pirzada AS, Alsharif KF, Daglia M, Khan H. Computational pharmacology and computational chemistry of 4-hydroxyisoleucine: Physicochemical, pharmacokinetic, and DFT-based approaches. Front Chem. 2023; 11.
- Sarkar C, Das B, Rawat VS, Wahlang JB, Nongpiur A, Tiewsoh I, et al. Artificial Intelligence and Machine Learning Technology Driven Modern Drug Discovery and Development. Vol. 24, International Journal of Molecular Sciences. MDPI; 2023.
- Mignani S, Rodrigues J, Tomas H, Jalal R, Singh PP, Majoral JP, et al. Present drug-likeness filters in medicinal chemistry during the hit and lead optimization process: how far can they be simplified? Vol. 23, Drug Discovery Today. Elsevier Ltd; 2018. p. 605– 15.
- Chagas CM, Moss S, Alisaraie L. Drug metabolites and their effects on the development of adverse reactions: Revisiting Lipinski’ s Rule of Five. Vol. 549, International Journal of Pharmaceutics. Elsevier B. V. ; 2018. p. 133– 49.
- Zhou SF, Zhong WZ. Drug design and discovery: Principles and applications. Vol. 22, Molecules. MDPI AG; 2017.
- Sliwoski G, Kothiwale S, Meiler J, Lowe EW. Computational methods in drug discovery. Vol. 66, Pharmacological Reviews. 2014. p. 334– 95.
- Carpenter KA, Cohen DS, Jarrell JT, Huang X. Deep learning and virtual drug screening. Vol. 10, Future Medicinal Chemistry. Future Medicine Ltd. ; 2018. p. 2557– 67.
- Meyer JG, Liu S, Miller IJ, Coon JJ, Gitter A. Learning Drug Functions from Chemical Structures with Convolutional Neural Networks and Random Forests. J Chem Inf Model. 2019;
- Patel L, Shukla T, Huang X, Ussery DW, Wang S. Machine Learning Methods in Drug Discovery. Molecules. 2020 Nov 2; 25 (22).
- Vamathevan J, Clark D, Czodrowski P, Dunham I, Ferran E, Lee G, et al. Applications of machine learning in drug discovery and development. Vol. 18, Nature Reviews Drug Discovery. Nature Publishing Group; 2019. p. 463– 77.
- Taye MM. Understanding of Machine Learning with Deep Learning: Architectures, Workflow, Applications and Future Directions. Vol. 12, Computers. MDPI; 2023.
- Vo AH, Van Vleet TR, Gupta RR, Liguori MJ, Rao MS. An Overview of Machine Learning and Big Data for Drug Toxicity Evaluation. Chem Res Toxicol. 2020 Jan 21; 33 (1): 20– 37.
- Mahesh B. Machine Learning Algorithms-A Review. International Journal of Science and Research [Internet]. 2018; Available from: https://www. researchgate. net/publication/344717762
- Nasteski V. An overview of the supervised machine learning methods. HORIZONSB. 2017 Dec 15; 4: 51– 62.
- Lavecchia A. Machine-learning approaches in drug discovery: Methods and applications. Vol. 20, Drug Discovery Today. Elsevier Ltd; 2015. p. 318– 31.
- Yosipof A, Guedes RC, Garcí a-Sosa AT. Data mining and machine learning models for predicting drug likeness and their disease or organ category. Front Chem. 2018 May 1; 6 (MAY).
- Dara S, Dhamercherla S, Jadav SS, Babu CM, Ahsan MJ. Machine Learning in Drug Discovery: A Review. Artif Intell Rev. 2022 Mar 1; 55 (3): 1947– 99.
- Singh A. ‘ Bharati Vidyapeeth’ s Institute of Computer Applications and Management (BVICAM). ’ 2016.
- Stå hl N, Falkman G, Karlsson A, Mathiason G, Boströ m J. Deep Convolutional Neural Networks for the Prediction of Molecular Properties: Challenges and Opportunities Connected to the Data. J Integr Bioinform. 2018 Dec 5; 16 (1).
- Alghushairy O, Ali F, Alghamdi W, Khalid M, Alsini R, Asiry O. Machine learning-based model for accurate identification of druggable proteins using light extreme gradient boosting. J Biomol Struct Dyn. 2023;
- Wen N, Liu G, Zhang J, Zhang R, Fu Y, Han X. A fingerprints based molecular property prediction method using the BERT model. J Cheminform. 2022 Dec 1; 14 (1).
- Hammann F, Gutmann H, Vogt N, Helma C, Drewe J. Prediction of adverse drug reactions using decision tree modeling. Clin Pharmacol Ther. 2010 Jul 24; 88 (1): 52– 9.
- Shi H, Liu S, Chen J, Li X, Ma Q, Yu B. Predicting drug-target interactions using Lasso with random forest based on evolutionary information and chemical structure. Genomics. 2019 Dec 1; 111 (6): 1839– 52.
- Sugaya N, Ikeda K. Assessing the druggability of protein-protein interactions by a supervised machine-learning method. BMC Bioinformatics. 2009 Aug 25; 10: 263.
- Tian S, Wang J, Li Y, Li D, Xu L, &, Hou T. The application of in silico drug-likeness predictions in pharmaceutical research. Adv Drug Deliv Rev. 2015; 86: 2– 10.
- Rashid M, Afzal O, Saleh A, Altamimi A. Benzimidazole Molecule Hybrid with Oxadiazole Ring as Antiproliferative Agents: In-Silico Analysis, Synthesis, and Biological Evaluation. Vol. 66, J. Chil. Chem. Soc. 2021.
- Ibrahim ZY, Uzairu A, Shallangwa GA, Abechi SE. Pharmacokinetic predictions and docking studies of substituted aryl amine-based triazolopyrimidine designed inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase (PfDHODH). Futur J Pharm Sci. 2021 Dec; 7 (1).
- Atkinson H, Mahon-Smith K, Elsby R. Drug Permeability and Transporter Assessment: Polarized Cell Lines. In: The ADME Encyclopedia. Springer International Publishing; 2021. p. 1– 13.
- Daina A, Michielin O, Zoete V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017 Mar 3; 7.
- Liu M, Zhang L, Li S, Yang T, Liu L, Zhao J, et al. Prediction of hERG potassium channel blockage using ensemble learning methods and molecular fingerprints. Toxicol Lett. 2020 Oct 10; 332: 88– 96.
- Kwon S, Bae H, Jo J, Yoon S. Comprehensive ensemble in QSAR prediction for drug discovery. BMC Bioinformatics. 2019 Oct 26; 20 (1).
- Janardhanan Y, Prathyush S, Chitesh K, Bhavesh P. Drug Classification and Repurposing Using Decision Tree Algorithm and Data Analysis. Int J Res Appl Sci Eng Technol. 2024 Apr 30; 12 (4): 461– 5.
- Qi Y. Random Forest for Bioinformatics. Ensemble Machine Learning. 2012; 307– 23.
- Jia CY, Li JY, Hao GF, Yang GF. A drug-likeness toolbox facilitates ADMET study in drug discovery. Vol. 25, Drug Discovery Today. Elsevier Ltd; 2020. p. 248– 58.
- Fatima S, Hussain A, Amir S Bin, Ahmed SH, Aslam SMH. XGBoost and Random Forest Algorithms: An In-Depth Analysis. Pakistan Journal of Scientific Research, PJOSR. 2023; 3 (1): 26– 31.
- Sagi O, Rokach L. Ensemble learning: A survey. Vol. 8, Wiley Interdisciplinary Reviews: Data Mining and Knowledge Discovery. Wiley-Blackwell; 2018.
- Chen T, Guestrin C. XGBoost: A Scalable Tree Boosting System. In: Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. New York, NY, USA: ACM; 2016. p. 785– 94.
- Brown LR. Biomaterials in Their Role in Creating New Approaches for the Delivery of Drugs, Proteins, Nucleic Acids, and Mammalian Cells in Safety Pharmacology. In: Drug Discovery and Evaluation: Safety and Pharmacokinetic Assays. Cham: Springer International Publishing; 2022. p. 1– 27.
- Ciobanu MM, Manoliu DR, Ciobotaru MC, Anchidin BG, Matei M, Munteanu M, et al. The Influence of Sensory Characteristics of Game Meat on Consumer Neuroperception: A Narrative Review. Foods. 2023 Mar 22; 12 (6): 1341.