
MINI REVIEW
Quality by Design: Approach to Analytical Method Validation
Academic Editor: Garnadi Jafar
Sciences of Pharmacy|Vol. 1, Issue 1, pp. 27-33 (2022)
CC BY 4.0-2022 Authors
Views
Downloads
Shares
Received
May 31, 2022Revised
Jun 18, 2022Accepted
Jun 22, 2022Published
Jun 27, 2022
Abstract
A pharmaceutical industry is highly regulated by a quality policy in its management. The principles of Quality by Design (QbD) must be applied to ensure the development of pragmatic and systematic methods while managing the risks associated with analytical methods. Quality by Design (QbD) is a scientific way to develop easy and robust analytical techniques for critical analysis. Quality by Design (QbD) is a systematic approach to product or method development that starts with predetermined goals and uses a science and risk management approach to achieve product and method understanding. The concept of risk management is deeply integrated into the quality assurance system to ensure pharmaceutical quality and patient safety. In the context of quality control, detecting impurities in raw materials and finished products is a major concern. Analytical Quality by Design (AQbD) aims to achieve quality in measurement. The main objectives are to explain the various steps involved in developing a method using a Quality by Design (QbD) approach for the development of analytical methods and to explain the implementation of Quality by Design (QbD) in the validation of analytical procedures. The advantages of applying Quality by Design (QbD) principles to analytical techniques include finding and minimizing sources of variability that could lead to poor method robustness and ensuring that the method meets the desired performance requirements over the product and method life cycle. The Analytical Quality by Design (AQbD) strategy is increasingly being adopted as it allows an early understanding of the method and guarantees the determination of a wider set of experimental conditions.
Introduction
Dr Joseph M. Juran first developed the concept of Quality by Design (QbD) in various publications. Dr Joseph M. Juran mentions that quality can be planned. The ICH Q8 guideline mentions the concept of QbD, which states, "Quality must be built into the product by design, but quality cannot be tested into the product". Quality is the suitability of a drug compound or drug for its intended use. The term includes attributes such as identity, purity, and strength. ICH Q8 Quality by Design is “ a systematic approach to development that begins with a defined goal and emphasizes product and process understanding and process control, based on knowledge and quality risk management". Quality in measurement is the main goal of QbD analytics (1). QbD principles as risk and knowledge-based decisions, a systematic approach to the development process, and continuous improvement lead to a "capable" process (2). The benefit of applying Analytical Quality by Design (AqbD) is understanding method attributes. Improved knowledge sharing, method development, and dynamic control strategy lead to more efficient regulatory oversight, operation elasticity, scientific and rational regulatory filing, shortens product time to be on the market, limited product rejection and reduces change after approval. Analytical methods are a fundamental part of the control strategy in pharmaceutical quality systems (ICH Q10). It includes many parameters and attributes related to medicinal compounds and products, i. e. operating conditions of the instruments and associated methods (3).
On par with the QbD process, the outcomes of AQbD are well understood and suitable for their intended purpose of robustness throughout the lifecycle. Lifecycle AQbD has different tools such as ATP (Analytical Target Profile), CQA Risk Assessment, Optimization and Method Development with DoE, MODR (method operable design region), Control Strategy and Risk Assessment, Validation of AQbD Method, and Continuous Method Monitoring. Graphical abstract showing the AQbD lifecycle with each tool (3).
Analytical Target Profile
Another approach to QbD analytics involves the definition of an analytical target profile (ATP), the set of criteria that define what is to be measured and the criteria for the performance of the method. Using material, process, and product attributes as inputs, ATP thus defines the analytical variables to be measured (e. g. a certain level of impurities) and the performance characteristics to be obtained by these measurements (e. g. accuracy, precision and distance). ATP presents the relationship between the final analytical method and the chemical process or formulation. As a chemical process is defined, for example, the need for a specific analytical method to measure impurities at a given level can be defined in ATP. Various methods can then be developed to satisfy ATP throughout the development cycle; for example, a generic method could be used early in development to meet ATP. A more efficient method that satisfies the original ATP could be developed at the product launch. The main driving factors behind the ATP's approach are the desire to gain flexibility in the methods used to perform the assays and to avoid costly post-approval changes to registered analytical methods that require proactive discussions with regulatory bodies. On the other hand, in this scenario, each ATP-compliant method will be properly validated and documented but will not require changes to the registered methods, as only ATP will be registered (4).
Analytical procedures must be demonstrated to be fit for purpose, which applies to their entire life cycle. A QbD approach has been proposed for pharmaceutical analysis to achieve this goal, including three stages of Procedure Design and Development, Procedure Performance Qualification, and Advanced Procedure Performance Verification according to process validation in the intended manufacturer (5).
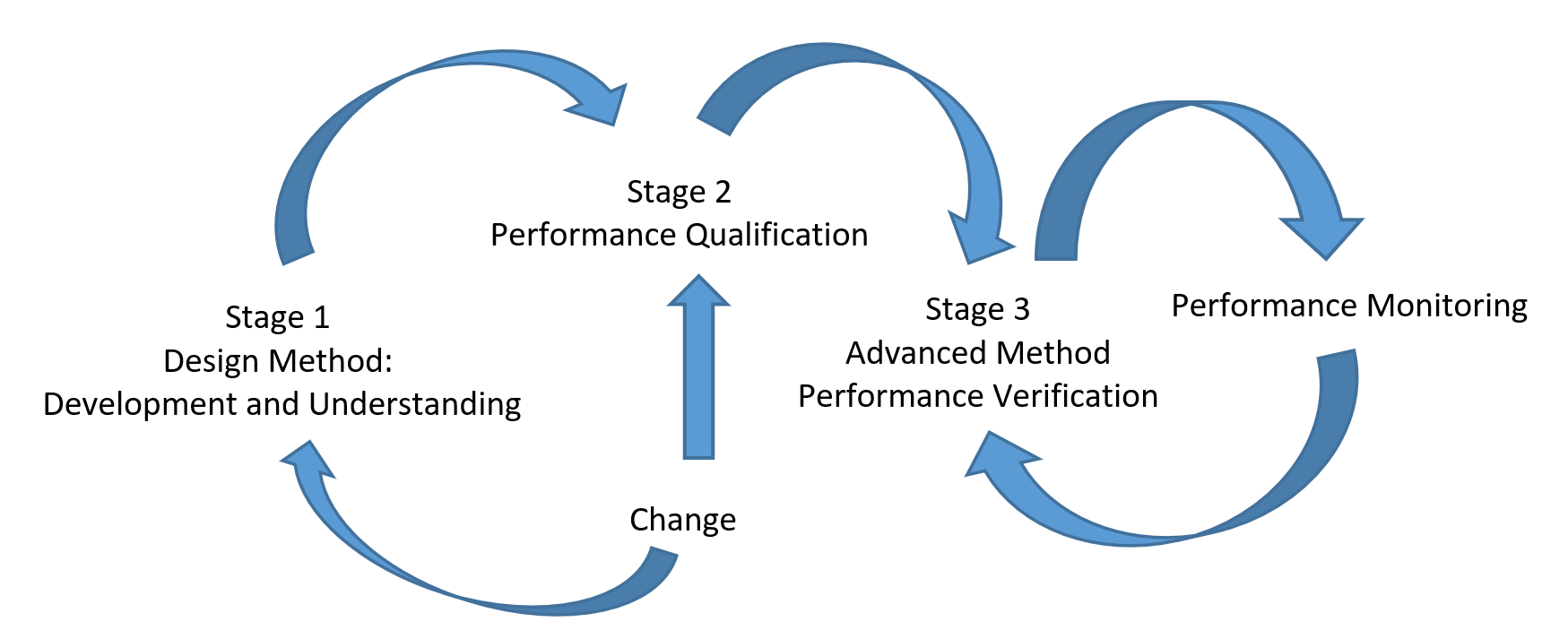
Identification of ATP includes selecting method requirements such as target analytes (products and impurities), analytical technique category, and product specification. An initial risk assessment will be conducted to anticipate method requirements and analytical criticality. Typical ATP for analytical procedures are as follows:
- Selection of target analytes (API and impurity),
- Selection of techniques (HPLC, GC, HPTLC, Ion Chromatography, Chiral HPLC, etc. ),
- Selection of method requirements (test profile, impurities, solvent residue) (6).
Critical Quality Attributes (CQA)
CQA is defined as physical, chemical, biological, or microbiological properties or characteristics that must be within the appropriate limits, ranges, or distributions to ensure the desired product quality (7, 8). CQA can also be interpreted as product properties that must be kept within a certain range/limit to achieve a reasonable shelf life (9). Critical process parameters may be the type of equipment, batch size, mixing order, mixing time, other operational conditions, etc. In addition, important raw material attributes are quality and quantity. These conditions must be within limits, range or distribution to ensure quality (8). CQA for analytical methods includes method attributes and method parameters. The performance characteristics of the method can be assessed through the parameters presented in Table 1. Each analytical technique has a different CQA. HPLC (UV or RID) CQA is a mobile phase buffer, pH, diluent, column selection, organic modifier, and elution method. GC method, Its CQA is gas flow, temperature and oven, injection temperature, diluent sample, and concentration. HPTLC method, the CQA is TLC plate, mobile phase, injection concentration and volume, plate development time, color development reagent, and detection method. Natural impurities and DS can determine CQA for developing analytical methods such as solubility, pH value, polarity, charged functional groups, boiling point, and solution stability (10). Determining CQA is very important in the QBD approach, as they have the greatest influence on final product quality and performance characteristics (which affect safety and efficacy) and needs to be controlled (11). Identification of CQA attributes is an important step in developing biopharmaceuticals that depend on a thorough understanding of the potential as a quality attribute by influencing safety and efficacy. The CQA assessment depends on the needs of the product attributes necessary for the expected product performance as defined by the quality target product profile (QTPP) and considers other sources of information for consideration in the next step (12).
| Performance characteristics | Definition | Category |
|---|---|---|
| Accuracy | The closeness of the results obtained with the actual value | Systematic variability |
| Specificity | Ability to rate the analyte against other components that might be expected to be present | |
| Linearity | The ability to obtain test results directly or by a well-defined mathematical transformation proportional to the concentration of the analyte in the sample within a certain range | |
| Precision | The ability of an analytical method to show the closeness of a series of measurements obtained from a homogeneous sample. | Inherent random variability |
| Limit of detection (LOD) | Limit test characteristics: lowest amount of analyte in the sample that can be detected. | |
| Limit of quantitation(LOQ) | The lowest amount of analyte in a sample can be determined with acceptable precision and accuracy. | |
| Range | The interval between the indicated upper and lower analyte levels is determined with the appropriate level of precision, accuracy and linearity. | - |
| Robustness | The capacity to remain unaffected by small but deliberate variations of procedural parameters listed in the procedure documentation indicates their conformity over the normal range. | - |
Risk Assessment
Risk assessment is a scientific process under quality risk management; it facilitates identifying which material attributes and process parameters could potentially affect product CQA. After the parameters are identified, mathematical tools are used to achieve a higher level of process understanding (13). Once CQA has been studied, the next step is to describe the relevant risk assessment. Once the technique is identified, QbD analytics focuses on assessing the risks associated with variability, including the analysis method, instrument configuration, measurement and method parameters, sample characteristics, sample preparation, and environmental conditions (14). Quality risk management and pharmaceutical quality system approaches are published in the relevant ICH guidelines (Q9 and Q10) (15).
According to the ICH Q9 guidelines, risk assessment is a systematic process for assessing, controlling, communicating and reviewing quality risks throughout the product life cycle. Prior product knowledge, such as accumulated laboratory, non-clinical and clinical experience with certain product quality attributes, is key in risk assessment. Risk identification, risk analysis, and risk evaluation are the three steps of risk assessment (7, 16). The steps taken in identifying risks can be seen in Figure 2.
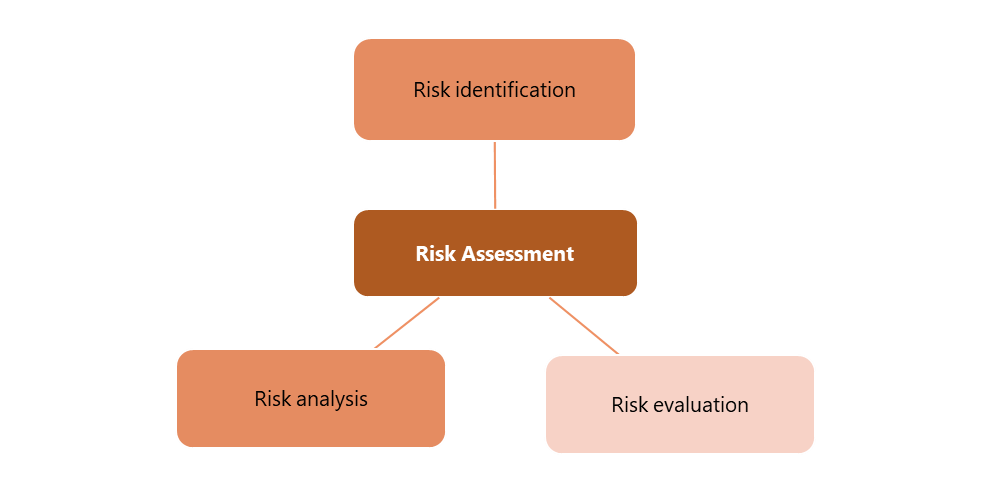
The first step of a risk assessment is critical to identifying and prioritizing potential risks. These risks include the instrument's operating method, reagent characteristics and cycle times. It is most desirable to specify an alternative method if the primary method fails. Flow charts and checklists are used to describe risk factors. The second step of risk assessment is risk evaluation. Fishbone diagrams are used to perform risk assessments, also called Ishikawa. According to this approach, risk factors are divided into three categories: high risk, noise, and experimental. After conducting a risk analysis, a risk evaluation is carried out. This risk assessment is designed to evaluate the knowledge of the attributes and uncertain processes and impacts. Later, this series of assessments produce recommendations that are under the results of an understanding of risk (17).
DoE: Design of Experiments (Method Optimization and Development)
Experimental design (DoE) is one of the most important steps of AQbD. DoE is a set of statistical tools that includes filtering and design optimization, which can provide better results with fewer trials. The screening design allows one to identify the most relevant independent variable (e. g. analytical condition) affecting the dependent variable (e. g. chromatographic response). In addition, the optimization design makes it possible to construct mathematical relationships that describe the dependent variable as a function of the independent variables, including the linear, quadratic, and interaction terms of the regression equation (18).
In the DOE approach, different types of objectives can be considered. Among these are response surface methodologies based on empirical models, which will predict outputs across the domains of interest (19). After the potential and critical analytical method variables are defined with an initial risk assessment, then DoE can be performed to confirm and correct the critical method variables based on statistical significance. Experimental Design (DOE) is a structured set of methods for determining the relationship between factors that affect the output of a process. One approach to the research characterization process is designed using an experimental design (DOE), so that data can be used to understand and define the design space, which is then executed, and the results are analyzed to determine the importance of the parameters and their role in building the design space (20). Can be specified per unit operation or a combination of several selected method variables and their interactions and responses (critical method attributes). This approach provides a good opportunity to filter out several conditions resulting from several experiments. Then, data evaluation using statistical tools is very important to identify the critical method variables and the appropriate optimal range for the method variables in which a robust area for critical method attributes can be obtained. Following the ICH Q8 guidelines, process robustness is defined as "The ability of a process to tolerate material variability and process and equipment changes without a negative impact on quality." The starting material's properties will affect the chemical process's toughness, impurity profile, physicochemical properties, processability, and drug stability. Understanding the process will provide sufficient knowledge to define resistance parameters by evaluating various operating conditions, scales, and equipment.
Method Operational Design Region (MODR)
The output of the DOE forms the basis for the MODR. MODR is the operating range for the critical method input variables (almost the same as CQA) that produces results that consistently meet the goals set in ATP. After the method development and risk assessment have been identified, the next step is the method operational design region. MODR is used as an operational area development method for day-to-day operations. The MODR is a science-based, risk-based, and multivariate approach to measuring the effect of various factors on method performance. It controls important methods such as system compliance, RRT and RRF. Following the ICHQ8 guidelines on "Design space" requirements in product development, the MODR can also be defined in the method development phase, which can serve as a source for robust and cost-effective methods (21).
Control Strategy
The control strategy is a control design tool. Controls can include parameters and attributes related to drug substances and ingredients and drug components, facilities and equipment operating conditions, in-process controls, finished product specifications and methods and frequency related to monitoring and control (20). It is calculated from the nature of the analyst and understanding of MODR. Strategy control methods can be set on the complete statistical data collected during the MODR. The control strategy is not always a one-time implementation carried out during the method development phase but can change with different phases of the method life cycle. It should be noted that the control strategy of the QbD approach is no different from the conventional approach (22). The control strategy can include three control levels: (i) Level 1, an automated control technique to monitor input material attributes and process parameters in real-time to ensure that CQA consistently complies with established acceptance criteria. (ii) Level 2 consists of pharmaceutical controls to reduce reliance on final product testing through flexible understanding of product material attributes and process parameters for identifying sources of variability impacting product quality within the defined design space. (iii) Level 3 is the control level traditionally used in the pharmaceutical industry (20).
Validation of AQbD Method
The AQbD method validation approach validates analytical methods on different API batches. This approach uses DoE and MODR knowledge to design method validation for all types of API build changes without re-validation. This approach provides the necessary elements of ICH validation and information about interactions, measurement uncertainty, control strategies, and continuous improvement. This approach requires fewer resources than traditional validation approaches without compromising quality (23). Therefore, the implementation of AQbD in the manufacturing process can be used as a control strategy to ensure the performance and quality of the specified product. This includes parameters and attributes related to medicinal substances and ingredients of medicinal products, including facilities, finished product specifications, instrument operating conditions, and associated approaches and frequencies (21).
Continuous Monitoring Method (CMM)
Life cycle management is a control strategy used to implement the design space at the commercial stage. The CMM is the final step in the AQbD life cycle. AQbD is an ongoing process of sharing knowledge acquired during the development and implementation of the design space. This includes the risk assessment results, assumptions based on prior knowledge, statistical design considerations, and bridging between the design space, MODR, control strategy, CQA, and ATP. Once method validation is complete, the method can be used for routine purposes and monitor the method's ongoing performance. AQbD can be performed using control charts or tracking system compliance data, method-related investigations, etc. CMM enables analysts to proactively identify and address out-of-trend performance (23, 24).
Advantages and Recommendations
AQbD is an approach that goes from reactive problem solving to proactive failure reduction. The risk assessment type and level depending on the development timeline's project stage. The success rate of AQbD depends on the right approach, planning, use of tools, and work performance promptly. Implementing the right risk assessment tools at the right time can lead to method failure prevention and a better understanding of the design space and control strategy.
Conclusion
Recently, the Food Drug Administration (FDA) has approved the application of a new drug based on analytical QbD to establish a protocol for method transfer, a methodology for MODR validation, and determine review criteria for evaluating QbD-based analytical methods. Implementing AQbD provides a powerful analytical method, which plays an important role in drug product development. The AQbD strategy facilitates an in-depth understanding of risk management methods and management. Optimization of AQbD-compliant methods must account for prediction errors and their spread to manage risk. The QbD methodology promotes a better understanding of the material characteristics and process parameters that affect the final quality of the targeted product. It also brings a holistic, risk-based, structured way of thinking into manufacturing procedures. A risk-based approach, the QbD approach above, can cover the early phases of pharmaceutical research activities, with the advantage of being more time and cost-effective from the research phase to market approval and industrial-scale manufacturing. Implementation of QbD and AQbD includes harmonization of terminology and concepts, training and education of human resources for industry and regulatory agencies, and the need for guidance on the documentation of knowledge generated during pharmaceutical method development.
Declarations
Conflict of Interest
The authors declare no conflicting interest.
Data Availability
Not applicable.
Ethics Statement
Not applicable.
Funding Information
Not applicable.
References
- Article R. Quality By Design: A Systematic Approach for the Analytical Method. 2019;9:1006–12.
- Mahapatra A, Meyyanathan SN. Analytical Quality by Design – A Review. Indian Res J Pharm Sci. 2020;7(2):2132–40.
- Jayagopal B, Shivashankar M. Analytical Quality by Design – A Legitimate Paradigm for Pharmaceutical Analytical Method Development and Validation . Mech Mater Sci Eng J. 2017;9(April):10.
- Psimadas D, Georgoulias P, Valotassiou V, Loudos G. Molecular Nanomedicine Towards Cancer : J Pharm Sci. 2012;101(7):2271–80.
- Ermer J. Analytical Target Profile: establishment of precision requirements for assay. J Pharm Biomed Anal. 2018;160:73–9.
- Wu H, White M, Khan MA. Quality-by-Design (QbD): An integrated process analytical technology (PAT) approach for a dynamic pharmaceutical co-precipitation process characterization and process design space development. Int J Pharm. 2011;405(1–2):63–78.
- Patil AS, Pethe AM. Quality by design (QbD): A new concept for development of quality pharmaceuticals. Int J Pharm Qual Assur. 2013;4(2):13–9.
- Pramod K, Tahir Ma, Charoo N, Ansari S, Ali J. Pharmaceutical product development: A quality by design approach. Int J Pharm Investig. 2016;6(3):129.
- George M, Ghosh I. Identifying the correlation between drug/stabilizer properties and critical quality attributes (CQAs) of nanosuspension formulation prepared by wet media milling technology. Eur J Pharm Sci. 2013 Jan;48(1–2):142–52.
- Raman NVVSS, Mallu UR, Bapatu HR. Analytical Quality by Design Approach to Test Method Development and Validation in Drug Substance Manufacturing. J Chem. 2015;2015:1–8.
- Mishra V, Thakur S, Patil A, Shukla A. Quality by design (QbD) approaches in current pharmaceutical set-up. Expert Opin Drug Deliv. 2018 Aug 3;15(8):737–58.
- Alt N, Zhang TY, Motchnik P, Taticek R, Quarmby V, Schlothauer T, et al. Determination of critical quality attributes for monoclonal antibodies using quality by design principles. Biologicals. 2016 Sep;44(5):291–305.
- Kalyan S, Vihar P. Quality by Design : Changing Outlook of Pharmaceutical Development. Int J Pharm Sci Res. 2019;10(9):4100–8.
- Gaykar D, Khadse SC. A review on analytical quality by design. Int J Pharm Sci Rev Res. 2017;44(2):96–102.
- Pallagi E, Ismail R, Paál TL, Csóka I. Initial Risk Assessment as part of the Quality by Design in peptide drug containing formulation development. Eur J Pharm Sci. 2018 Sep;122:160–9.
- Kasap D, Kaymak M. Risk Identification Step of the Project Risk Management. In: PICMET ’07 - 2007 Portland International Conference on Management of Engineering & Technology. IEEE; 2007. p. 2116–20.
- Kelley B. Quality by Design risk assessments supporting approved antibody products. MAbs. 2016 Nov 16;8(8):1435–6.
- Moreira C dos S, Lourenço FR. Development and optimization of a stability-indicating chromatographic method for verapamil hydrochloride and its impurities in tablets using an analytical quality by design (AQbD) approach. Microchem J. 2020 May;154:104610.
- Myers RH, Montgomery DC. Response Surface Methodology. IIE Trans. 1996;28(12):1031–2.
- Yu LX, Amidon G, Khan MA, Hoag SW, Polli J, Raju GK, et al. Understanding Pharmaceutical Quality by Design. AAPS J. 2014 Jul 23;16(4):771–83.
- Ramalingam P, Jahnavi B. QbD Considerations for Analytical Development. In: Pharmaceutical Quality by Design. Elsevier; 2019. p. 77–108.
- Sangshetti JN, Deshpande M, Zaheer Z, Shinde DB, Arote R. Quality by design approach: Regulatory need. Arab J Chem. 2017 May;10:S3412–25.
- Scypinski S, Roberts D, Oates M, Etse J. Pharmaceutical Research and Manufacturers Association acceptable analytical practice for analytical method transfer. Pharm Technol. 2002;26(3):84–9.
- Snee R. Out-of-Control Signals. 2015;(November).