
RESEARCH ARTICLE
Phytocompound Inhibitors of Caspase-3 as a Beta-Cell Apoptosis Treatment Development Option: An In Silico Approach
Academic Editor: James H. Zothantluanga
Sciences of Phytochemistry|Vol. 2, Issue 1, pp. 14-30 (2023)
CC BY 4.0-2023 Authors
Views
Downloads
Shares
Received
Jan 18, 2023Revised
Feb 28, 2023Accepted
Mar 8, 2023Published
Mar 9, 2023
Abstract
The prevalence of Diabetes mellitus (DM) is continuously rising worldwide. Among its types, type I is characterized by the destruction of beta cells triggered by various mechanisms, including the activation of Caspase 3. Studies have demonstrated the crucial role of Caspase 3 in initiating the apoptosis of beta cells in DM. Our research aims to identify possible phytocompounds inhibitors of Caspase 3 using computational approach. We obtained 3D structures of Caspase 3 and 6511 phytocompounds from the Protein Data Bank and the African Natural Products Database, respectively. The phytocompounds were assessed for druglikeness properties, topological polar surface area, and preliminary toxicity using DataWarrior. The phytocompounds were subjected to molecular docking simulation (MDS) at Caspase 3 active site using AutoDock-Vina. The frontrunner phytocompounds obtained from the MDS were subjected to protease inhibition prediction on Molinspiration. The pharmacokinetics of the phytocompounds were assessed on SwissADME. The in-depth computational toxicity profile of the phytocompounds was evaluated on the pkCSM web. The binding interactions of the phytocompounds with Caspase 3 were assessed with Discovery Studio Visualizer and Maestro. Seventeen phytocompounds were found to have no violation of Lipinski's rule and had no toxicity based on the preliminary assessment, have better binding affinity and protease inhibitory prediction scores than the references, have optimistic bioactivity radar prediction and similar amino acids interaction, in comparison with the references. Further studies, which include in-vitro and in-vivo studies, will be carried out to validate the results of this study.
Introduction
Beta-cell apoptosis is a critical event in the pathogenesis of type 1 diabetes mellitus (DM). Aside from being the primary mechanism by which cells are destroyed, beta-cell apoptosis has been linked to the onset of type 1 DM via antigen cross-presentation mechanisms that result in beta-cell-specific T-cell activation (1). Apoptosis can be activated via the extrinsic death receptor or intrinsic mitochondrial pathway, activating effector caspases (2). Apoptosis is also a critical process in the development of atherosclerosis (2).
Caspases are endoproteases and genes crucial for preserving homeostasis by controlling cell death and inflammation. A phylogenetically conserved death program that is essential for the homeostasis and growth of higher organisms carefully regulates their activation. Numerous human diseases are primarily pathogenetic due to the dysregulation of apoptosis. Caspases are potential therapeutic targets because they are part of the apoptotic machinery (3, 4).
Caspases are classified broadly according to their known roles in apoptosis (caspase-3, -6, -7, -8, and -9 in mammals) and inflammation (caspase-1, -4, -5, -12 in humans and caspase-1, -11, and -12 in mice). Caspase-2, -10, and -14 functions are more difficult to classify. Caspases involved in apoptosis have been divided into two groups based on their mechanism of action: initiator caspases (caspases - 8 and -9) and executioner caspases (caspase-3, -6, and -7) (3).
In a study titled "Caspase-3-Dependent -Cell Apoptosis in the Initiation of Autoimmune Diabetes Mellitus", the authors used a genetic approach to show that this process is necessary for cross-presentation of beta-cell antigen to activate beta-cell-specific T cells (1). They proved that mice lacking caspase-3 do not experience the onset of autoimmune diabetes, which is indicated by normal level of glucose concentration in the blood, unaffected beta-cells revealing high insulin content, and absence of beta-cell specific T-cell activation in the pancreatic draining lymph nodes. In a different study titled "Immunocytochemical localization of caspase-3 in pancreatic islets from type 2 diabetic subjects", the author reported finding more cleaved caspase-3 immunostained islets from type 2 diabetics, which may indicate an accelerated apoptotic cascade in the islets, along with increasing amyloid deposition before ultimate cell death (5).
The improper control of caspase-mediated cell death and inflammation is linked to various illnesses, including inflammatory, neurological, and other metabolic diseases and cancer. It may be necessary to therapeutically target caspase-3 activity in cells to stop the onset of autoimmune diabetes (1). Numerous natural and synthetic caspase inhibitors have been discovered and created to be used therapeutically. Only a few synthetic caspase inhibitors have progressed into clinical trials due to their lacklustre efficacy or harmful side effects. They have yet to prove compelling enough for patient use (6).
The aim of this study is to detect phytocompounds with drug like properties in African plants that could inhibit Caspase-3 through in silico analysis.
Materials and Methods
Materials
The materials used are personal computer, African Natural Compounds Database, PubChem (https://pubchem. ncbi. nlm. nih. gov) (7), Linux operating system (Ubuntu desktop 18.04), Protein data bank (https://www. rcsb. org/) (8), DataWarrior software (9), PyMOL software (10), AutoDockTools-1.5.6 software (11), AutoDock Vina 1.1.2 software (12), on Ubuntu operating system, and Molinspiration Chemoinformatics web tool (https://www. molinspiration. com/cgi-bin/properties) (13).
Literature Mining
To find essential targets and receptors for apoptotic processes, literatures were explored. This was done to examine the role of the target and receptors in the pathophysiology and initiation of cell apoptosis. This provides more details regarding the receptor's characteristics, activities, and druggability.
Selection and Preparation of the Receptor
Caspase 3 in 3D format was retrieved from Protein Data Bank (PDB) with the PDB ID: 3KJF after various targets and receptors had been identified, literature had been mined, and the target and receptor had been analyzed. PyMOL program was initially used to prepare the pdb file by selecting the necessary chains and deleting multiple ligands. To understand how the ligands attach to receptors, PyMOL software was used. The AutoDockTools was used to get the receptor ready for molecular docking simulations. The receptors were prepared by adding polar hydrogens and Kollman's charges before storing them in the pdbqt file format, the structural format needed for performing molecular docking simulation on Autodock vina. As shown in Table 1, the electrostatic grid boxes and the three-dimensional affinity with various sizes and centers were formed around the protein's active region.
| Axis | Centers | Sizes |
|---|---|---|
| X | 21.94 | 14 |
| Y | -4.306 | 14 |
| Z | 10.718 | 14 |
Selection of the Ligands
In this study, 6511 phytocompounds were examined, which were obtained from the African Natural Products Database (african-compounds. org) (14, 15). The compounds were downloaded as 3D-structure data files for analysis. Various parameters such as partition coefficient (Log P), topological polar surface area (TPSA), molecular weight, hydrogen bond donor, and hydrogen bond acceptor were used to assess the phytocompounds. Some of the phytocompounds were found to infringe Lipinski's rule. Those that did not breach the rule underwent toxicological assessment for mutagenicity, carcinogenicity, tumorigenicity, and reproductive effect.
Preparation of the Ligands
Phytocompounds with no Lipinski’s rule of five infarction and no predicted toxicity (mutagenicity, carcinogenicity, tumorigenicity, and reproductive effect) in silico were prepared for the molecular docking simulation. Reference ligands were identified from the literature, including the compound co-crystallized with the receptor/protein on the PDB database. In preparation for the ligands for molecular docking simulation, all rotatable bonds, torsions, and Gasteiger charges were assigned and saved in the pdbqt file format.
Validation of Docking Protocol
The PDB structure of the 3KJF (Caspase 3) protein, in association with a reference inhibitor as was downloaded from the PDB, was replicated in silico to validate the molecular docking simulations procedure for this protein. Other known inhibitors of Caspase 3 were also used for the validation, including Flubendazole, Fenoprofen, Pranoprofen, and Diflunisal (16). The AutoDockTools-1.5.6 was used to calculate polar hydrogen, Kollman charges, grid box sizes, and centers at a grid space of 1.0 (11, 12). The protein was stored as a pdbqt file. AutoDockTools-1.5.6 was used to prepare the reference chemicals for molecular docking simulation. Torsion-free bonds, as well as any other rotatable bonds, were permitted. After that, files with the pdbqt extension were generated as output. On a Linux environment, a virtual screening shell script was used to locally implement the AutoDockVina® molecular docking simulation of the protein and reference chemical utilizing the centers and sizes (12). Co-crystal inhibitor binding interaction was compared with the re-docked co-crystalized compounds, Flubendazole, Fenoprofen, Pranoprofen, and Diflunisal using PyMol-1.4.1 software and Discovery studio visualizer.
Molecular Docking of the Phytocompounds on Caspase 3
The phytocompounds were prepared in batches for molecular docking simulations using virtual screening scripts against the Caspase 3. Following the validation of docking methods, four replicates of Molecular Docking Simulations were performed on a Linux platform using AutoDockVina® and related tools. To determine the leading phytocompounds, binding free energy values (kcal/mol SD) were ranked.
Bioactivity Prediction of Phytocompounds
The online Molinspiration web tool version 2011.06 (www. molinspiration. com) was supplied SMILES notations of the leading phytocompounds to forecast the bioactivity scores for protease inhibition.
Computational Pharmacokinetics of Frontrunner Phytocompounds
The top phytocompounds underwent a thorough pharmacokinetics evaluation using SwissADME, a web-based tool that assesses the druglikeness, physicochemical, ADME properties, and medicinal chemistry compatibility of small molecules (17). The assessment was conducted to examine the pharmacokinetics of the lead phytocompounds in detail.
In-depth Toxicity Assessment of Frontrunner Phytocompounds
An in-depth toxicity prediction of the frontrunner phytocompounds for AMES toxicity, Max. tolerated dose (human), hERG I inhibitor, hERG II inhibitor, Oral Rat Acute Toxicity (LD50), Oral Rat Chronic Toxicity (LOAEL), Hepatotoxicity, Skin Sensitization, T. Pyriformis toxicity and Minnow toxicity on the pkCSM platform (18).
Analysis of the Frontrunner Phytocompounds-Caspase 3 Binding Interactions
The amino acids of Caspase 3 binding interactions with each frontrunner phytocompounds were analyzed using Discovery Studio Visualizer v20.1.0.19295, and Maestro 13.3 aided the generation of 2D structures of the interaction for easy observation (19, 20).
Results
Preliminary Drug-likeness and Toxicity Assessment of the Ligands (Phytocompounds)
The drug-likeness assessment of the 6511 phytocompounds was performed using Lipinski's rule of five to screen out phytocompounds that violated the guidelines on the DataWarrior application. Following the screening, 3814 phytocompounds had no infraction of Lipinski's rule, but 2697 phytocompounds did. Toxicity testing on the 3814 phytocompounds that did not violate Lipinski's criteria was performed using DataWarrior to discover phytocompounds that could be mutagenic, tumorigenic, irritating, or have reproductive implications. In silico testing revealed that 1897 phytocompounds possessed none of the identified toxicities. The total polar surface area (TPSA) was also calculated for each phytocompounds.
Validation of Docking Protocol
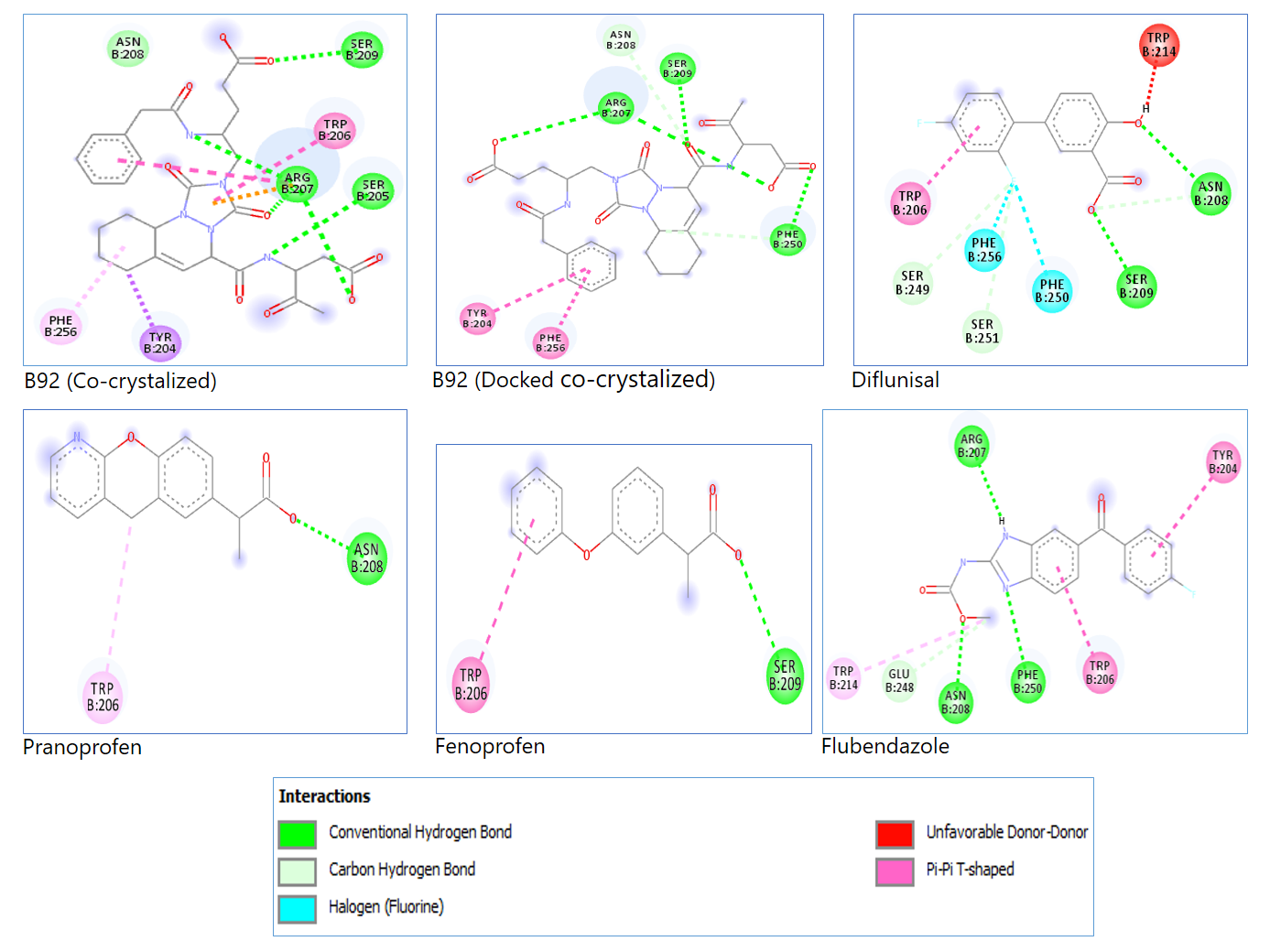
The docking procedure was validated to assure the in silico repeatability of the experimental protein-ligand interactions gathered from the protein data bank and to observe Caspase 3 amino-acids-conventional hydrogen bond interactions with the reference compounds known as inhibitors of caspase 3. This validation ensures accuracy, reliability, and reproducibility in computational predictions. Figure 1 is the 2D representation of the docked co-crystalized ligand and reference compounds with the specific Caspase 3 amino acids involved in the interaction, visually illustrating key molecular interactions. Table 2 shows the binding energy of the docked co-crystalized ligand and that of the reference known inhibitors of caspase 3, providing a comparative assessment of their binding affinities. Table 3 shows each reference compound, docked co-crystalized ligand, and the docked co-crystalized ligand with the specific amino acids involved in their interaction with caspase 3, further detailing binding site preferences and interaction strength.
| No. | Reference compounds | Mean Binding Affinity | Standard Deviation |
|---|---|---|---|
| 1 | Flubendazole | -7.60 | 0.20 |
| 2 | Diflunisal | -7.20 | 0.00 |
| 3 | B92 (Co-crystalized) | -7.13 | 0.15 |
| 4 | Pranoprofen | -6.50 | 0.00 |
| 5 | Fenoprofen | -6.18 | 0.05 |
| No. | Reference compounds | Amino acids |
|---|---|---|
| 1 | B92 | ARG 207, SER 205, SER 209 |
| 2 | B92 (Co-crystalized) | ARG 207, SER 209, PHE 250 |
| 3 | Diflunisal | ASN 208, SER 209 |
| 4 | Pranoprofen | ASN 208 |
| 5 | Fenoprofen | SER 209 |
| 6 | Flubendazole | ARG 207, ASN 208, PHE 250 |
Molecular Docking of the Phytocompounds against Caspase 3
To identify phytocompounds with greater in silico binding energies against Caspase 3 than the co-crystalized ligand and reference compounds, molecular docking of the phytocompounds was carried out on Caspase 3. The result is presented in Table 4, showing phytocompounds with higher mean binding energies than the co-crystalized ligand and reference compounds. The table also contains Lipinski's rule parameters and TPSA values of the phytocompounds.
| No. | Compound name | Mean Binding Affinity | Standard Deviation (±) | Molecular Weight | Octanol-Water Coefficient | Hydrogen Bond Acceptors | HydrogenDonor | TopologicalPolar Surface Area |
|---|---|---|---|---|---|---|---|---|
| 1 | Amataine | -9.20 | 0.00 | 493.65 | -2.75 | 7.00 | 2.00 | 49.04 |
| 2 | 3'-epi-afroside | -9.20 | 0.00 | 534.64 | 1.07 | 9.00 | 4.00 | 134.91 |
| 3 | Neoilexonol | -8.70 | 0.00 | 442.73 | 6.67 | 2.00 | 1.00 | 37.30 |
| 4 | Chrysophanol-10, 10'-bianthrone | -8.70 | 0.00 | 478.50 | 4.70 | 6.00 | 4.00 | 115.06 |
| 5 | Hydroxyhopane | -8.50 | 0.00 | 426.73 | 7.16 | 1.00 | 1.00 | 20.23 |
| 6 | Taraxast-20-ene-3beta, 30-diol | -8.50 | 0.00 | 446.76 | 8.49 | 2.00 | 2.00 | 40.46 |
| 7 | Caulindole A | -8.50 | 0.00 | 368.52 | 5.38 | 2.00 | 2.00 | 31.58 |
| 8 | Acacic acid lactone | -8.50 | 0.00 | 470.69 | 4.79 | 4.00 | 2.00 | 66.76 |
| 9 | 3-oxo-12beta-hydroxy-Oleanan-28, 13beta-olide | -8.50 | 0.00 | 430.67 | 5.60 | 3.00 | 1.00 | 46.53 |
| 10 | Lucidene | -8.50 | 0.00 | 416.60 | 8.00 | 2.00 | 0.00 | 18.46 |
| 11 | Millettone | -8.40 | 0.00 | 382.41 | 1.24 | 6.00 | 0.00 | 77.05 |
| 12 | Taraxasterol | -8.30 | 0.00 | 424.71 | 7.00 | 1.00 | 1.00 | 20.23 |
| 13 | 5, 6-dehydrocalotropin | -8.30 | 0.00 | 532.63 | 0.79 | 9.00 | 3.00 | 131.75 |
| 14 | Chrysophanol- isophyscionBianthrone | -8.30 | 0.00 | 508.53 | 4.63 | 7.00 | 4.00 | 124.29 |
| 15 | Uguenensene | -8.30 | 0.00 | 484.59 | 2.99 | 7.00 | 0.00 | 87.50 |
| 16 | Calotroproceryl acetate A | -8.10 | 0.00 | 466.75 | 7.74 | 2.00 | 0.00 | 26.30 |
| 17 | Lupeol | -8.10 | 0.00 | 440.75 | 7.98 | 1.00 | 1.00 | 20.23 |
| 18 | Beta-amyrin | -8.10 | 0.00 | 426.73 | 7.34 | 1.00 | 1.00 | 20.23 |
| 19 | Anastatin B | -8.10 | 0.00 | 378.34 | 3.58 | 7.00 | 4.00 | 120.36 |
| 20 | 3-hydroxycycloart-24-one | -8.10 | 0.00 | 442.73 | 6.86 | 2.00 | 1.00 | 37.30 |
| 21 | Diketo leucolactone | -8.10 | 0.00 | 468.68 | 5.04 | 4.00 | 1.00 | 63.60 |
| 22 | Sigmoidin E | -8.08 | 0.22 | 406.48 | 5.55 | 5.00 | 2.00 | 75.99 |
| 23 | Di-podocarpanoid hugonone A | -8.08 | 0.25 | 586.85 | 4.53 | 6.00 | 5.00 | 118.22 |
| 24 | 24-methylene cycloartanol | -8.05 | 0.06 | 440.75 | 8.34 | 1.00 | 1.00 | 20.23 |
| 25 | Isojamaicin | -8.05 | 0.06 | 378.38 | 3.73 | 6.00 | 0.00 | 63.22 |
| 26 | Seneganolide | -8.03 | 0.05 | 470.52 | 1.37 | 8.00 | 1.00 | 112.27 |
| 27 | 24-methylencycloartanol | -8.00 | 0.00 | 438.74 | 8.08 | 1.00 | 1.00 | 20.23 |
| 28 | Scalarolide | -8.00 | 0.00 | 386.57 | 4.51 | 3.00 | 1.00 | 46.53 |
| 29 | Citriquinochroman | -8.00 | 0.00 | 442.47 | 3.88 | 7.00 | 2.00 | 89.79 |
| 30 | Matricolone | -8.00 | 0.00 | 286.41 | 3.36 | 2.00 | 1.00 | 37.30 |
| 31 | Epi-lupeol | -8.00 | 0.00 | 426.73 | 7.65 | 1.00 | 1.00 | 20.23 |
| 32 | 20-epi-isoiguesterinol | -8.00 | 0.00 | 424.62 | 5.19 | 3.00 | 2.00 | 57.53 |
| 33 | Melliferone | -8.00 | 0.00 | 452.68 | 5.64 | 3.00 | 0.00 | 43.37 |
| 34 | Abyssinone I | -8.00 | 0.00 | 322.36 | 3.87 | 4.00 | 1.00 | 55.76 |
| 35 | Argeloside O | -7.93 | 0.05 | 521.63 | 0.31 | 9.00 | 0.00 | 112.58 |
| 36 | Calotropursenyl acetate B | -7.90 | 0.00 | 468.76 | 7.84 | 2.00 | 0.00 | 26.30 |
| 37 | Beta-anhydroepidigitoxigenin | -7.90 | 0.00 | 356.50 | 3.47 | 3.00 | 1.00 | 46.53 |
| 38 | 3-acetyltaraxasterol | -7.90 | 0.00 | 468.76 | 7.84 | 2.00 | 0.00 | 26.30 |
| 39 | Lupeol acetate | -7.90 | 0.00 | 480.77 | 8.20 | 2.00 | 0.00 | 26.30 |
| 40 | Siphonellinol C | -7.90 | 0.00 | 490.72 | 5.04 | 5.00 | 4.00 | 90.15 |
| 41 | Isoadiantol | -7.90 | 0.00 | 426.73 | 7.11 | 1.00 | 1.00 | 20.23 |
| 42 | 3-acetylsesterstatin 1 | -7.90 | 0.00 | 446.63 | 4.07 | 5.00 | 1.00 | 72.83 |
| 43 | 1, 5-di-O-caffeoylquinic acid | -7.90 | 0.00 | 426.73 | 7.59 | 1.00 | 0.00 | 17.07 |
| 44 | Tingenin B | -7.90 | 0.00 | 438.61 | 4.55 | 4.00 | 2.00 | 74.60 |
| 45 | Friedelane-3, 7-dione | -7.90 | 0.00 | 440.71 | 6.88 | 2.00 | 0.00 | 34.14 |
| 46 | Norisojamicin | -7.90 | 0.00 | 364.35 | 3.46 | 6.00 | 1.00 | 74.22 |
| 47 | A-homo-3a-oxa-5beta-Olean-12-en-3- one-28-oic acid | -7.90 | 0.00 | 471.70 | 3.58 | 4.00 | 0.00 | 66.43 |
| 48 | Corosolic acid | -7.90 | 0.00 | 473.72 | 3.18 | 4.00 | 2.00 | 80.59 |
| 49 | Lupenone | -7.88 | 0.05 | 424.71 | 7.79 | 1.00 | 0.00 | 17.07 |
| 50 | Coladin | -7.88 | 0.17 | 424.54 | 4.92 | 5.00 | 0.00 | 61.83 |
| 51 | Abyssinone III | -7.88 | 0.19 | 390.48 | 5.90 | 4.00 | 1.00 | 55.76 |
| 52 | Neomacrotriol | -7.85 | 0.06 | 472.75 | 6.63 | 3.00 | 3.00 | 60.69 |
| 53 | Abyssinoflavone V | -7.85 | 0.06 | 338.36 | 3.53 | 5.00 | 2.00 | 75.99 |
| 54 | 13-hydroxyfeselol | -7.85 | 0.06 | 400.51 | 3.53 | 5.00 | 2.00 | 75.99 |
| 55 | Assafoetidnol A | -7.83 | 0.05 | 398.50 | 3.15 | 5.00 | 2.00 | 75.99 |
| 56 | Demethoxyexcelsin | -7.83 | 0.05 | 384.38 | 3.15 | 7.00 | 0.00 | 64.61 |
| 57 | 3-taraxasterol | -7.80 | 0.00 | 430.76 | 9.48 | 1.00 | 1.00 | 20.23 |
| 58 | Neoilexonol acetate | -7.80 | 0.00 | 484.76 | 7.16 | 3.00 | 0.00 | 43.37 |
| 59 | Sipholenol I | -7.80 | 0.00 | 508.74 | 3.59 | 6.00 | 4.00 | 102.68 |
| 60 | Cabralealactone | -7.80 | 0.00 | 412.61 | 5.00 | 3.00 | 0.00 | 43.37 |
| 61 | Ursolic acid | -7.80 | 0.00 | 455.70 | 3.76 | 3.00 | 1.00 | 60.36 |
| 62 | Stylopine | -7.80 | 0.00 | 328.39 | 0.46 | 5.00 | 2.00 | 44.66 |
| 63 | Khayanolide D | -7.80 | 0.00 | 502.56 | 1.07 | 9.00 | 3.00 | 135.66 |
| 64 | Olean-12-en-3-one | -7.80 | 0.00 | 426.73 | 7.59 | 1.00 | 0.00 | 17.07 |
| 65 | Tribulus saponin aglycone 1 | -7.80 | 0.00 | 350.54 | 4.75 | 3.00 | 2.00 | 49.69 |
| 66 | Foetidin | -7.80 | 0.00 | 381.49 | 5.47 | 4.00 | 2.00 | 51.83 |
| 67 | Samarcandin | -7.80 | 0.00 | 400.51 | 3.47 | 5.00 | 2.00 | 75.99 |
| 68 | Resinone | -7.80 | 0.00 | 440.71 | 6.94 | 2.00 | 1.00 | 37.30 |
| 69 | Uncinatone | -7.80 | 0.00 | 318.41 | 3.97 | 4.00 | 2.00 | 66.76 |
| 70 | Urs-9 (11), 12-dien-3beta-ol | -7.80 | 0.14 | 424.71 | 7.17 | 1.00 | 1.00 | 20.23 |
| 71 | Sesamin | -7.78 | 0.05 | 354.36 | 3.22 | 6.00 | 0.00 | 55.38 |
| 72 | Euphornin C | -7.75 | 0.30 | 546.70 | 4.95 | 8.00 | 1.00 | 116.20 |
| 73 | Salmahyrtisol B | -7.75 | 0.06 | 386.57 | 4.51 | 3.00 | 1.00 | 46.53 |
| 74 | Isoferprenin | -7.75 | 0.06 | 362.47 | 6.42 | 3.00 | 0.00 | 35.53 |
| 75 | (±)-paulownia | -7.75 | 0.06 | 370.36 | 2.40 | 7.00 | 1.00 | 75.61 |
| 76 | Limonin | -7.75 | 0.06 | 470.52 | 1.03 | 8.00 | 0.00 | 104.57 |
| 77 | Sablacaurin A | -7.73 | 0.15 | 482.79 | 9.30 | 2.00 | 0.00 | 26.30 |
| 78 | Farnesiferol A | -7.73 | 0.05 | 384.51 | 3.58 | 4.00 | 1.00 | 55.76 |
| 79 | Epilupeol | -7.70 | 0.00 | 426.73 | 7.65 | 1.00 | 1.00 | 20.23 |
| 80 | Lupenone | -7.70 | 0.00 | 424.71 | 7.79 | 1.00 | 0.00 | 17.07 |
| 81 | Sipholenol A | -7.70 | 0.00 | 478.76 | 5.38 | 4.00 | 3.00 | 69.92 |
| 82 | Taraxasteryl acetate | -7.70 | 0.00 | 468.76 | 7.84 | 2.00 | 0.00 | 26.30 |
| 83 | Retusolide B | -7.70 | 0.00 | 316.44 | 2.95 | 3.00 | 0.00 | 43.37 |
| 84 | Cycloart-23Z-ene-3beta, 25-diol | -7.70 | 0.00 | 456.75 | 7.32 | 2.00 | 1.00 | 29.46 |
| 85 | 7-deacetoxy-7-oxogedunin | -7.70 | 0.00 | 440.53 | 2.82 | 6.00 | 0.00 | 86.11 |
| 86 | Tribulus saponin aglycone 2 | -7.70 | 0.00 | 434.66 | 4.18 | 4.00 | 3.00 | 69.92 |
| 87 | Lup-20 (29)-ene-3beta, 23-diol | -7.70 | 0.00 | 456.75 | 7.05 | 2.00 | 2.00 | 40.46 |
| 88 | Beta-boswellic acid | -7.70 | 0.00 | 455.70 | 3.93 | 3.00 | 1.00 | 60.36 |
| 89 | 3-ketotirucall-8, 24-dien-21-oic acid | -7.70 | 0.00 | 425.63 | 4.43 | 3.00 | 0.00 | 57.20 |
| 90 | 6-oxoisoiguesterin | -7.70 | 0.00 | 420.59 | 6.02 | 3.00 | 2.00 | 57.53 |
| 91 | Friedelanol methyl ether | -7.70 | 0.00 | 470.82 | 8.44 | 1.00 | 0.00 | 9.23 |
| 92 | Jamaicin | -7.70 | 0.00 | 378.38 | 3.73 | 6.00 | 0.00 | 63.22 |
| 93 | Calopogonium isoflavone B | -7.70 | 0.00 | 348.35 | 3.80 | 5.00 | 0.00 | 53.99 |
| 94 | Di-podocarpanoid hugonone B | -7.70 | 0.00 | 580.80 | 4.03 | 6.00 | 4.00 | 115.06 |
| 95 | 3-O-benzoylhosloquinone | -7.70 | 0.00 | 420.55 | 5.24 | 4.00 | 0.00 | 60.44 |
| 96 | Isochamanetin | -7.68 | 0.05 | 364.40 | 3.80 | 5.00 | 3.00 | 86.99 |
| 97 | Oleanolic acid | -7.68 | 0.05 | 457.72 | 4.09 | 3.00 | 1.00 | 60.36 |
| 98 | Pectachol B | -7.68 | 0.05 | 442.55 | 4.29 | 6.00 | 1.00 | 74.22 |
| 99 | 3-O-benzoylhosloppone | -7.65 | 0.10 | 420.55 | 4.76 | 4.00 | 1.00 | 63.60 |
| 100 | Lup-20 (29)-ene-3-acetate | -7.63 | 0.05 | 467.76 | 5.87 | 2.00 | 0.00 | 40.13 |
| 101 | Marmaricin | -7.63 | 0.05 | 384.51 | 3.93 | 4.00 | 1.00 | 55.76 |
| 102 | Calactin | -7.60 | 0.00 | 532.63 | 0.79 | 9.00 | 3.00 | 131.75 |
| 103 | Olibanumol H | -7.60 | 0.00 | 460.74 | 5.66 | 3.00 | 3.00 | 60.69 |
| 104 | Botulin | -7.60 | 0.00 | 442.73 | 6.72 | 2.00 | 2.00 | 40.46 |
| 105 | Proscillaridin | -7.60 | 0.00 | 532.67 | 2.09 | 8.00 | 4.00 | 125.68 |
| 106 | Ottelione B | -7.60 | 0.00 | 312.41 | 3.93 | 3.00 | 1.00 | 46.53 |
| 107 | Sesterstatin 7 | -7.60 | 0.00 | 444.61 | 4.14 | 5.00 | 1.00 | 72.83 |
| 108 | Sonchuside A | -7.60 | 0.00 | 416.51 | 1.08 | 8.00 | 4.00 | 125.68 |
| 109 | 3alpha-acetoxyolean-12-en-28-al | -7.60 | 0.00 | 499.75 | 4.58 | 4.00 | 0.00 | 66.43 |
| 110 | Beta-amyrin acetate | -7.60 | 0.00 | 468.76 | 7.65 | 2.00 | 0.00 | 26.30 |
| 111 | Isoiguesterin | -7.60 | 0.00 | 408.62 | 5.84 | 2.00 | 1.00 | 37.30 |
| 112 | 5beta, 24-cyclofriedelan-3-one | -7.60 | 0.00 | 424.71 | 7.29 | 1.00 | 0.00 | 17.07 |
| 113 | Sigmoidin B | -7.60 | 0.00 | 356.37 | 3.83 | 6.00 | 4.00 | 107.22 |
| 114 | Sigmoidin F | -7.60 | 0.00 | 422.48 | 5.20 | 6.00 | 3.00 | 96.22 |
| 115 | 3'-prenylnaringenin | -7.60 | 0.00 | 338.36 | 4.36 | 5.00 | 3.00 | 86.99 |
| 116 | Abyssinin I | -7.60 | 0.00 | 368.38 | 3.46 | 6.00 | 2.00 | 85.22 |
| 117 | Durmillone | -7.60 | 0.00 | 378.38 | 3.73 | 6.00 | 0.00 | 63.22 |
| 118 | Hugonone A | -7.60 | 0.00 | 584.84 | 4.64 | 6.00 | 4.00 | 115.06 |
| 119 | 3-oxo-12-oleanen-28-oic acid | -7.60 | 0.00 | 453.68 | 4.13 | 3.00 | 0.00 | 57.20 |
| 120 | Limonyl acetate | -7.60 | 0.00 | 514.57 | 1.37 | 9.00 | 0.00 | 113.80 |
| 121 | Flubendazole | -7.60 | 0.20 | |||||
| 122 | Diflunisal | -7.20 | 0.00 | |||||
| 123 | B92 | -7.13 | 0.15 | |||||
| 124 | Pranoprofen | -6.50 | 0.00 | |||||
| 125 | Fenoprofen | -6.18 | 0.05 |
Bioactivity Prediction of Phytocompounds
Because Caspase 3 is a protease, the phytocompounds were screened computationally on Molinspiration for their ability to inhibit protease. According to the results, 80 phytocompounds were discovered to have computational protease inhibitory scores below the co-crystalized ligand (B92) and above the four other reference compounds used (Flubendazole, Pranoprofen, Fenoprofen, and Diflunisal). Table 5 below shows the outcomes of the bioactivity prediction of the phytocompounds with higher binding energies than the co-crystalized ligand and reference compounds. One of the phytocompounds, as shown in Table 5, has a bioactivity score above Pranoprofen, Fenoprofen, and Diflunisal but below B92 and Flubendazole.
| No. | Phytochemical | Protease Inhibitory score | Plant source |
|---|---|---|---|
| 1 | B92 | 0.50 | |
| 2 | 9R-hydroxysarcophine | 0.41 | Sarcophyton glaucum |
| 3 | Beta-boswellic acid | 0.33 | Boswellia species |
| 4 | Sipholenol I | 0.3 | Callyspongia siphonella |
| 5 | Olean-12-en-3- one-28-oic acid | 0.28 | Albizia gummifera |
| 6 | Tribulus saponin aglycone 2 | 0.26 | Tribulus species |
| 7 | Urs-12-ene-1beta, 3beta, 11alpha, 15alpha-tetraol | 0.25 | Salvia argentea var. aurasiaca |
| 8 | Neoilexonol | 0.23 | Boswellia carterii |
| 9 | Ursolic acid | 0.23 | Amaracus akhdarensis |
| 10 | 3beta-hydroxy-11alpha-methoxyurs-12-ene | 0.22 | Launaea arborescens |
| 11 | 1, 5-di-O-caffeoylquinic acid | 0.21 | Cynara cardunculus |
| 12 | Olibanumol H | 0.21 | Boswellia carterii |
| 13 | Isoadiantol | 0.18 | Adiantum capillus-veneris |
| 14 | Lup-20 (29)-ene-3beta, 23-diol | 0.18 | Salvia palaestina |
| 15 | Uguenensene | 0.17 | Vepris uguenensis |
| 16 | (+)-7alpha, 8beta-dihydroxydeepoxysarcophine | 0.17 | Sarcophyton auritum |
| 17 | Neoilexonol acetate | 0.17 | Boswellia carterii |
| 18 | Cycloart-23Z-ene-3beta, 25-diol | 0.17 | Euphorbia bupleuroides |
| 19 | Taraxasterol | 0.16 | Calotropis procera |
| 20 | Lupeol | 0.16 | Salvia palaestina |
| 21 | Taraxasterol | 0.16 | Calotropis procera |
| 22 | Sonchuside A | 0.16 | Launaea arborescens |
| 23 | 3-O-alpha-L-arabinopyranosyl-echinocystic acid | 0.15 | Dizygotheca kerchoveana |
| 24 | Epilupeol | 0.15 | Boswellia species |
| 25 | Oleanolic acid | 0.15 | Salia triloba |
| 26 | Abyssinoflavone V | 0.14 | Erythrina abyssinica |
| 27 | Isoferprenin | 0.14 | Ferula communis var. genuina |
| 28 | Limonyl acetate | 0.14 | Vepris uguenensis |
| 29 | 3-hydroxycycloart-24-one | 0.13 | Euphorbia guyoniana |
| 30 | Sigmoidin E | 0.13 | Erythrina abyssinica |
| 31 | Tribulus saponin aglycone 1 | 0.13 | Tribulus species |
| 32 | Isochamanetin | 0.13 | Uvaria lucida ssp. lucida |
| 34 | Hydroxyhopane | 0.12 | Azolla nilotica |
| 35 | Siphonellinol C | 0.12 | Callyspongia siphonella |
| 36 | Urs-9 (11), 12-dien-3beta-ol | 0.12 | Boswellia carterii |
| 37 | Sipholenol A | 0.12 | Callyspongia siphonella |
| 38 | Calactin | 0.12 | Pergularia tomentosa |
| 39 | 3alpha-acetoxyolean-12-en-28-al | 0.12 | Salvia palaestina |
| 40 | Beta-amyrin | 0.11 | Trichodesma africanum |
| 41 | Abyssinone I | 0.11 | Erythrina abyssinica |
| 42 | Calotropursenyl acetate B | 0.11 | Calotropis procera |
| 43 | Lupeol acetate | 0.11 | Torilis radiata |
| 44 | Abyssinone III | 0.11 | Erythrina abyssinica |
| 45 | 3-acetylsesterstatin 1 | 0.1 | Hyrtios erecta |
| 46 | Sigmoidin F | 0.1 | Erythrina abyssinica |
| 47 | Resinone | 0.09 | Drypetes gerrardii |
| 48 | Euphornin C | 0.09 | Euphorbia helioscopia |
| 49 | Lucidene | 0.08 | Uvaria species |
| 50 | Calotroproceryl acetate A | 0.08 | Calotropis procera |
| 51 | Beta-anhydroepidigitoxigenin | 0.08 | Calotropis procera |
| 52 | 3-taraxasterol | 0.08 | Pergularia tomentosa |
| 53 | 3'-epi-afroside | 0.07 | Gomphocarpus sinaicus |
| 54 | Taraxast-20-ene-3beta, 30-diol | 0.07 | Launaea arborescens |
| 55 | 5, 6-dehydrocalotropin | 0.07 | Gomphocarpus sinaicus |
| 56 | Argeloside O | 0.07 | Solenostemma argel |
| 57 | Khayanolide D | 0.07 | Khaya senegalensis |
| 58 | 5beta, 24-cyclofriedelan-3-one | 0.07 | Drypetes gerrardii |
| 59 | 24-methylene cycloartanol | 0.06 | Euphorbia helioscopia |
| 60 | 24-methylencycloartanol | 0.06 | Euphorbia bupleuroides |
| 61 | Limonin | 0.06 | Vepris glomerata |
| 62 | Sesterstatin 7 | 0.06 | Hyrtios erecta |
| 63 | Beta-amyrin acetate | 0.06 | Scorzonera undulata |
| 64 | Anastatin B | 0.05 | Anastatica hierochuntica |
| 65 | Scalarolide | 0.05 | Hyrtios erecta |
| 66 | Retusolide B | 0.05 | Euphorbia retusa |
| 67 | 3-O-benzoylhosloquinone | 0.05 | Hoslundia opposita |
| 68 | Lup-20 (29)-ene-3-acetate | 0.05 | Euphorbia helioscopia |
| 69 | Neomacrotriol | 0.04 | Neoboutonia macrocalyx |
| 70 | 3-acetyltaraxasterol | 0.03 | Pergularia tomentosa |
| 71 | Tingenin B | 0.03 | Elaeodendron schlechteranum |
| 72 | Friedelane-3, 7-dione | 0.03 | Drypetes gerrardii |
| 73 | Taraxasteryl acetate | 0.03 | Achillea fragrantissima |
| 74 | Abyssinin I | 0.03 | Erythrina abyssinica |
| 75 | 3-oxo-12-oleanen-28-oic acid | 0.03 | Ekebergia benguelensis |
| 76 | Di-podocarpanoid hugonone A | 0.01 | Hugonia busseana |
| 77 | 20-epi-isoiguesterinol | 0.01 | Salacia madagascariensis |
| 78 | Lupenone | 0.01 | Diospyros mespiliformis |
| 79 | Sablacaurin A | 0.01 | Sabal causiarum |
| 80 | Lupenone | 0.01 | Diospyros mespiliformis |
| 81 | Hugonone A | 0.01 | Hugonia castaneifolia |
| Flubendazole | 0.01 | ||
| 82 | 7-deacetoxy-7-oxogedunin | 0.00 | Swietenia mahogani |
| Pranoprofen | -0.05 | ||
| Fenoprofen | -0.07 | ||
| Diflunisal | -0.14 |
Computation Pharmacokinetics of Frontrunner Phytocompounds
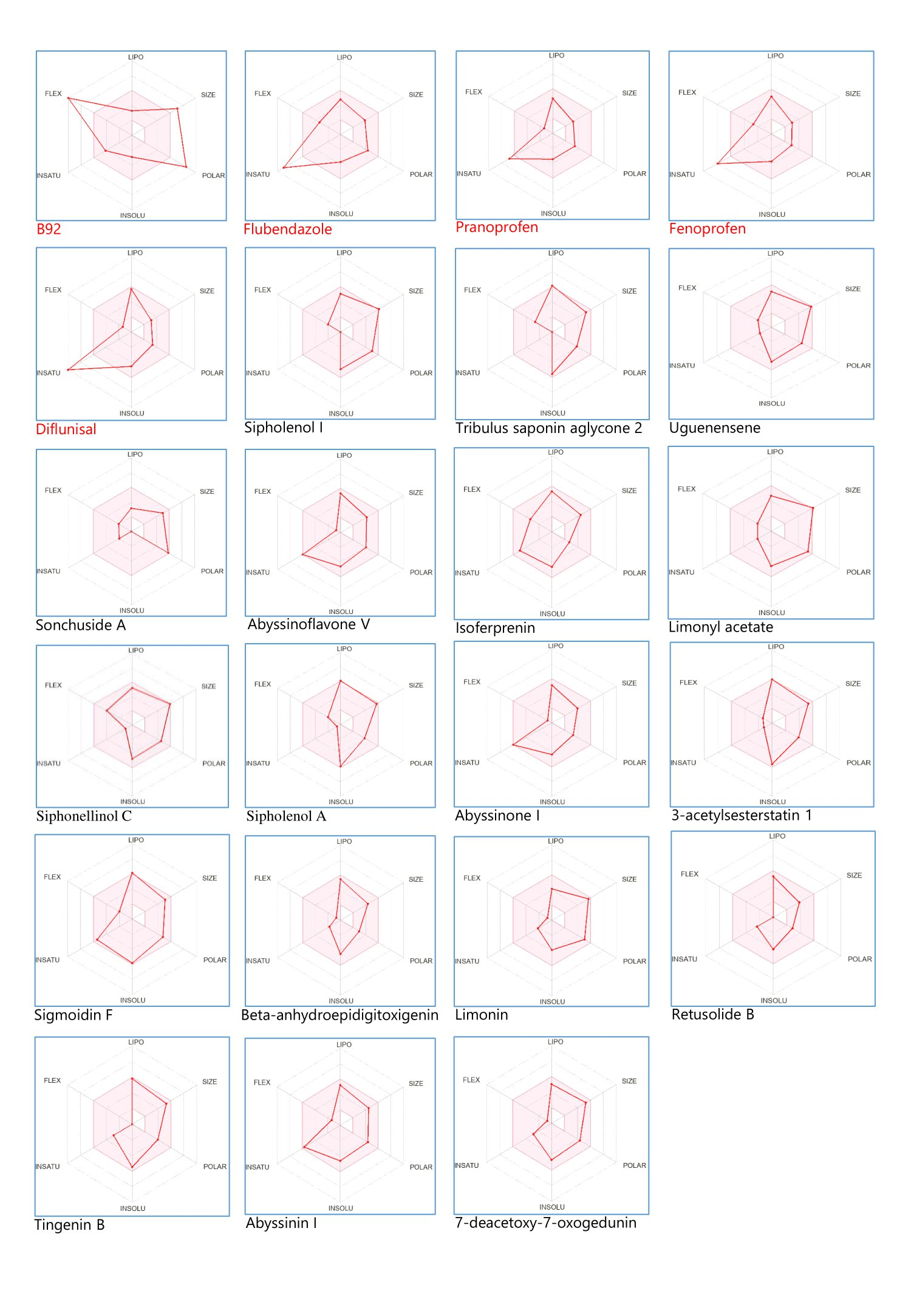
The pharmacokinetic assessment of frontrunner phytocompounds, reference compounds, and co-crystalized ligands is shown in Figure 2 using bioavailability radar graphics. The components include Lipophilicity (LIPO), size, polarity (POLAR), solubility (INSOLU), flexibility (FLEX), and saturation (INSATU). The reference compounds and co-crystalized ligands failed the bioavailability radar test. Of 82 phytocompounds assessed, 18 fell within the optimal bioavailability range.
In-depth Toxicity Prediction of Frontrunner Phytocompounds
The results of the in-depth toxicity prediction of the frontrunner compounds are presented in Table 6, showing different toxicities against which the phytocompounds were predicted.
| No. | Phytocompounds | AMES toxicity | Max. tolerated dose (human) | hERG I inhibitor | hERG II inhibitor | Oral Rat Acute Toxicity (LD50) | Oral Rat Chronic Toxicity | Hepatotoxicity | Skin Sensitization | T. Pyriformis toxicity | Minnow toxicity |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Tribulus saponin aglycone 2 | No | -0.925 | No | No | 2.220 | 1.629 | No | No | 0.396 | 0.289 |
| 2 | Uguenensene | No | -0.768 | No | No | 2.985 | 0.033 | No | No | 0.29 | 0.869 |
| 3 | Sonchuside A | No | 0.501 | No | No | 2.559 | 2.631 | No | No | 0.286 | 3.134 |
| 4 | Abyssinoflavone V | No | -0.245 | No | No | 2.475 | 1.863 | No | No | 0.609 | 1.939 |
| 5 | Isoferprenin | No | 0.482 | No | No | 2.329 | 2.227 | Yes | No | 1.462 | -2.800 |
| 6 | Limonyl | No | -0.773 | No | No | 3.604 | 1.195 | Yes | No | 0.286 | 1.963 |
| 7 | Siphonellinol C | No | -0.955 | No | No | 2.993 | -0.097 | No | No | 0.495 | 1.376 |
| 8 | Sipholenol A | No | -1.055 | No | Yes | 2.364 | 1.489 | No | No | 0.521 | 0.310 |
| 9 | Abyssinone I | Yes | -0.139 | No | No | 2.363 | 1.754 | No | No | 1.230 | 1.270 |
| 10 | 3-acetylsesterstatin 1 | No | -1.115 | No | No | 2.669 | 0.463 | No | No | 0.381 | -0.243 |
| 11 | Sigmoidin F | No | -0.267 | No | Yes | 2.274 | 2.036 | No | No | 0.332 | 0.285 |
| 12 | Beta-anhydroepidigitoxigenin | No | -0.567 | No | No | 2.001 | 1.726 | Yes | No | 0.637 | -0.142 |
| 13 | Limonin | No | -0.651 | No | No | 3.23 | 1.872 | No | No | 0.287 | 0.424 |
| 14 | Retusolide B | No | -0.032 | No | No | 1.868 | 1.665 | No | No | 0.657 | -0.738 |
| 15 | Tingenin B | No | -0.538 | No | No | 3.096 | 1.601 | Yes | No | 0.386 | 0.564 |
| 16 | Abyssinin I | No | 0.127 | No | No | 2.350 | 2.070 | No | No | 0.338 | 0.393 |
| 17 | 7-deacetoxy-7-oxogedunin | No | -0.793 | No | No | 2.598 | 1.760 | No | No | 0.321 | 0.638 |
| 18 | Flubendazole | Yes | 0.328 | No | Yes | 2.471 | 2.254 | Yes | No | 0.285 | 0.821 |
| 19 | Pranoprofen | No | 0.701 | No | No | 2.659 | 1.384 | Yes | No | 0.289 | 1.475 |
| 20 | Fenoprofen | No | 0.648 | No | No | 2.113 | 2.010 | No | No | 0.286 | 0.088 |
| 21 | Diflunisal | No | 0.956 | No | No | 2.789 | 2.443 | No | No | 0.286 | 1.357 |
| 22 | B92 | No | 1.419 | No | No | 2.321 | 2.765 | Yes | No | 0.285 | 2.568 |
Analysis of the Frontrunner Phytocompounds-Caspase 3 Binding Interactions
The binding interactions between the frontrunner phytocompounds and Caspase 3 were analyzed using Maestro 13.3 and Discovery studio visualizer. The results are presented in Figure 3 and Table 7. The result shows the specific amino acids (with arrow) that contributed to the conventional hydrogen bond interaction between the frontrunner phytocompounds and Caspase 3.
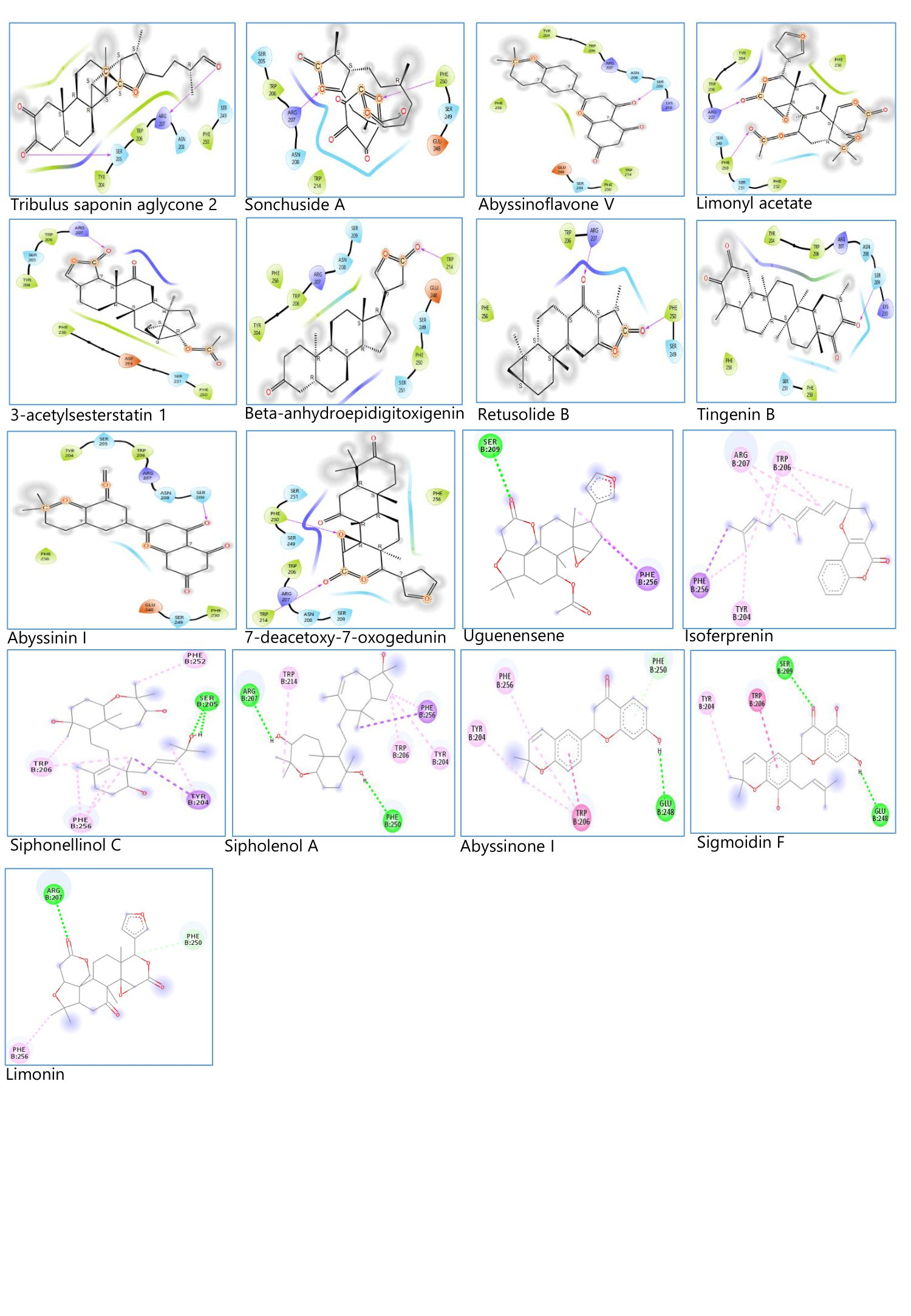
| No. | Frontrunner Phytocompounds | Amino Acids |
|---|---|---|
| 1 | Tribulus saponin aglycone 2 | ARG 207, SER 205 |
| 2 | Sonchuside | ARG 207, PHE 250 |
| 3 | Abyssinoflavone V | SER 209 |
| 4 | Limonyl | ARG 207, PHE 250 |
| 5 | 3-acetylsesterstatin 1 | ARG 207 |
| 6 | Beta-anhydroepidigitoxigenin | TRP 214 |
| 7 | Retusolide B | ARG 207, PHE 250 |
| 8 | Tingenin B | SER 209 |
| 9 | Abyssinin I | SER 209 |
| 10 | 7-deacetoxy-7-oxogedunin | PHE 250, TRP 214 |
| 11 | Uguenensene | SER 209 |
| 12 | Isoferprenin | - |
| 13 | Siphonellinol C | SER 205 |
| 14 | Sipholenol A | ARG 207, PHE 250 |
| 15 | Abyssinone I | GLU 248 |
| 16 | Sigmoidin F | SER 209, GLU 248 |
| 17 | Limonin | ARG 207 |
Discussion
The study aimed to use in silico molecular docking simulation to compare the binding affinities of phytocompounds from the African natural product database to the reference compounds against Caspase 3. The phytocompounds should have no violation of Lipinski's rule of five, with no predicted toxicity and positive bioactivity score. Due to the high cost of drug discovery and development and the required time, creating new medications has proven challenging. The "in silico" method of drug discovery and design, also known as computer-aided drug design, is now widely used in preliminary research to minimize the chances of compound failure in the later stages of drug development. Computer-aided drug design components like molecular docking, molecular dynamics, quantitative structure-activity relationship, absorption, distribution, metabolism, excretion, and toxicity tool and their precise predictions speed up the discovery and development of new drugs (21, 22).
On the other hand, medicines and medicinal substances have historically been derived from nature, primarily plants. Plant extracts have been evaluated for different pharmacological activities with promising results (23, 24). Most medicines today are either isolated or created from isolates derived from natural sources. Based on their use in conventional medical procedures, most currently utilized medications are made from natural sources (25). More novel compounds are being isolated from plants and deposited in chemical databases (26). There are also general biological and specialized databases on which thousands of proteins are deposited to aid scientific research (22).
During drug design, pharmaceutical chemists use Lipinski's rule of five to predict oral bioavailability. This study sourced 6,511 phytocompounds from African plants via the African natural product database. Drug-likeness was assessed using DataWarrior based on Lipinski’s rule. A compound is likely orally effective if it meets these criteria: a) molecular weight under 500; b) octanol/water partition coefficient (Log P) under 5; c) no more than five hydrogen bond donors; d) no more than ten hydrogen bond acceptors (27-29). Of the 6,511 phytocompounds analyzed, 3,814 met all Lipinski’s rule criteria.
The DataWarrior application used for Lipinski's rule and preliminary toxicity assessment employs a precomputed collection of structural pieces that trigger toxicity alerts when discovered in the structures under investigation. To compile these fragment lists, all compounds from the Registry of Toxic Effects of Chemical Substances (RTECS) database known to be active in a specific toxicity class were thoroughly split (30). The phytocompounds were first severed during the process, with each rotating link leading to a set of core fragments. These were then used to reconstruct each substantial substructure of the parent molecule. A substructure search process was then used to determine the frequency of any fragment (core and created fragments) within all chemicals in that toxicity class. It also found these fragment frequencies in the structural data of over 3000 traded medications. Any fragment was considered a risk factor if it was commonly encountered as a substructure of dangerous chemicals but never or only infrequently in traded pharmaceuticals. This assumption was based on the view that most drugs sold are free of or have less toxic effects. Predicated on this described fragments exploration, 1897 phytocompounds had no in silico mutagenicity, tumorigenicity, irritant, or reproductive effects. These phytocompounds contained no fragments or fragments widely recognized to have any of the toxicities enumerated in the Registry of Toxic Effects of Chemical Substances.
As shown in Table 4, the molecular docking findings revealed 120 phytocompounds with higher binding affinity than the reference compounds. Lower binding affinity indicates improved ligand binding. The most significant magnitude negative value, representing the most positive conformation of the complex formed whenever the ligand invested efficiently binds with the protein's active site, determines the significance of binding affinity values. As can be seen, the mean binding affinity scores are negative. This happens because protein-ligand binding occurs solely when the free energy change is negative. The G-level difference between complexed and unconjugated free states is commensurate to the stability of the protein-ligand interaction. Once ∆G is minimal in the system, protein folding and protein-ligand binding occur (31, 32). As a result, negative ∆G scores indicate the stability of the arising complexes with the receptor molecules, a necessary feature of effective drugs (33).
Compounds with better binding affinities than the reference compounds used were subjected to molinspiration bioactivity prediction for protease inhibition since Caspase is a protease. The result of the bioactivity prediction presented in Table 5 shows that 82 phytocompounds out of 120 compounds analyzed are active as protease inhibitors based on the range of bioactivity score on the molinspiration platform. In molinspiration, biological activity is measured by a bioactivity score that is categorized as active (0.00 to 0.5), moderately active (0.00 to -0.5), and inactive (less than -0.5) (13).
Bioavailability Radar is displayed for a rapid appraisal of drug-likeness. Six physicochemical properties are considered: lipophilicity, size, polarity, solubility, flexibility, and saturation. A physicochemical range on each axis was defined by descriptors such as size, lipophilicity, H-bonding characteristics, rotatable bond, aromatic ring counts, etc. , and depicted as a pink area in which the radar plot of the molecule has to fall entirely to be considered druglike (34, 35). The results of the pharmacokinetic prediction of the frontrunner phytocompounds presented in Figure 2, as a bioavailability radar, revealed that only 17 out of 82 phytocompounds analyzed fit optimally into the pink region of the bioavailability radar map.
The 17 phytocompounds with optimal bioavailability radar prediction were subjected to further toxicity prediction on the pkCSM application. The result is presented in Table 6 with the predictions of the reference compounds too. Seven parameters were predicted; AMES toxicity, hERG I inhibitor, hERG II inhibitor, Hepatotoxicity, Minnow toxicity, T. Pyriformis toxicity, Max. tolerated dose (human), Oral Rat Acute Toxicity (LD50), and Oral Rat Chronic Toxicity. The Ames test is a procedure that uses the amino acids needed by the bacterial strains Salmonella typhimurium and Escherichia coli to identify mutations. This mutation test aims to find revertant bacteria that give the original bacteria its ability to synthesize an essential amino acid back. The revertant bacteria can still grow when the original strain's necessary amino acid is absent (36).
Ames test results have shown that it is susceptible to predicting carcinogens: 80% of Ames evaluation mutagens are also cancerous, while a negative outcome has no discriminatory significance (a chemical negative in Salmonella has the same possibility of being either a non-carcinogen, a non-genotoxic carcinogen, or a genotoxic carcinogen acting through a process not observed by the Ames test (37). In the creation of cardiac action potentials, HERG is crucial. Hence, QT prolongation and unexpected cardiac death are linked to hERG channel inhibition. Due to this significant outcome, evaluating hERG blockade by compounds during the early stages of drug discovery and development is crucial. Using various drug descriptors like log P and log S and modeling techniques like estate fingerprint, CDK fingerprint, and secondary structural fingerprint, this inhibitory effect can be predicted in silico (38). Minnow toxicity is a crucial foundation for risk and hazard analysis of substances in the aquatic system (39). The toxicity of Tetrahymena pyriformis is frequently used as a toxic end state. From the result in Table 6, Abyssinone I recorded positive for Ames toxicity with one of the reference compounds, Flubendazole. Sipholenol A and Sigmoidin F were predicted positive as hERG inhibitors and Flubendazole. Abyssinoflavone V, Isoferprenin, Beta-anhydroepidigitoxigenin, Tingenin B, Flubendazole, and Pranoprofen were predicted to possess hepatotoxic effects.
The conventional hydrogen bond interaction between the amino acids of Caspase and each of the frontrunner phytocompounds were analyzed as presented in Figure 3 and Table 7. Observation of the interactions of the reference compounds with Caspase 3, as shown in Figure 1 and Table 3, reveals the specific amino acids-conventional hydrogen bond interactions, which are probably responsible for the actions of these reference compounds. The amino acids include ARG 207, SER 205, SER 209, ASN 208, and PHE 250. Now, observation of Figure 3 and Table 7 also reveals the specific amino acids interaction of the frontrunner compound with Caspase 3, similar to those of the reference compounds, except Beta-anhydroepidigitoxigenin, Abyssinone I, 7-deacetoxy-7-oxogedunin, Sigmoidin F and Isoferprenin
Based on the simulation research design, the outcomes of in silico research translate well throughout in vitro or in vivo studies. Before synthetic chemistry synthesis, in silico techniques are frequently used to examine compound libraries' bioavailability, toxicity, and promising bioactivity (40). Similarly to that, we planned the design of the current in silico study to increase the likelihood of getting positive results in bioassays. One of the quickest and most accurate in silico methods for analyzing the molecular interactions and chemical bonding between a ligand and a protein is molecular docking (41). To observe and analyze the molecular interaction and ligand binding of compounds with studied biomarkers, molecular docking research, and in vivo studies are often combined (42-48). This discussion demonstrates the reliability of in silico drug discovery and development studies, supports and validates the methodology used in the current in silico study, and supports the notion that the African natural product database contains promising phytocompounds that could have the potential to act as Caspase 3 inhibitor.
Conclusion
Inhibitors of Caspase 3 can offer a remedy for the pharmaceutical intervention of beta-cell apoptosis in diabetes since options for treating beta-cell apoptosis are a significant therapeutic need. In this study, the findings imply that Tribulus saponin aglycone 2, Sonchuside, Abyssinoflavone V, Limonyl, 3-acetylsesterstatin 1, Retusolide B, Tingenin B, Abyssinin I, Uguenensene, Siphonellinol C, Sipholenol A, Sigmoidin F and Limonin and possibly their plant sources are candidates for further studies as Caspase 3 inhibitors. These phytocompounds are predicted to be druglike, with optimal pharmacokinetic parameters. The compounds possess better binding energies than the reference compounds used for the study. The phytocompounds also have similar conventional hydrogen bond interaction with Caspase 3 compared to the reference compounds. Validating this in silico work requires further thorough research using different models, such as in vitro and in vivo assays using the phytocompounds or extracts containing the phytocompounds. Restate the problem, summarize the paper, and discuss the implications and future research direction. This conclusion must address the objective of the study.
Declarations
Acknowledgment
The authors are grateful to the Principal Investigator (Prof. Ikemefuna Uzochukwu) of the Drug Design and Informatics Laboratory, Faculty of Pharmaceutical Sciences, Nnamdi Azikiwe University, Awka, Anambra State, Nigeria, for providing computational facilities. We appreciate the contribution of the CURIES research team of the Faculty of Pharmaceutical Sciences, Nnamdi Azikiwe University, Awka, to this ongoing research.
Conflict of Interest
The authors declare no conflicting interest.
Data Availability
The unpublished data is available upon request to the corresponding author.
Ethics Statement
Not applicable.
Funding Information
Not applicable.
References
- Liadis N, Murakami K, Eweida M, Elford AR, Sheu L, Gaisano HY, Hakem R, Ohashi PS, Woo M. Caspase-3-dependent beta-cell apoptosis in the initiation of autoimmune diabetes mellitus. Mol Cell Biol. (2005) 25(9):3620–9.
- Sun J, Singh P, Österlund J, Orho‐Melander M, Melander O, Engström G, et al. Hyperglycaemia‐associated Caspase‐3 predicts diabetes and coronary artery disease events. J Intern Med. (2021) 290(4):855–65.
- McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol. (2013) 5(4):a008656–a008656.
- Sadowski-Debbing K, Coy JF, Mier W, Hug H, Los M. Caspases--their role in apoptosis and other physiological processes as revealed by knock-out studies. Arch Immunol Ther Exp (Warsz). (2002) 50(1):19-34.
- Tomita T. Immunocytochemical localisation of caspase-3 in pancreatic islets from type 2 diabetic subjects. Pathology. (2010) 42(5):432–7.
- Dhani S, Zhao Y, Zhivotovsky B. A long way to go: caspase inhibitors in clinical use. Cell Death Dis. (2021) 12(10):949.
- Kim S, Chen J, Cheng T, Gindulyte A, He J, He S, et al. PubChem in 2021: new data content and improved web interfaces. Nucleic Acids Res. (2021) 49(D1):D1388–95.
- Berman HM. (2000) The Protein Data Bank. Nucleic Acids Res. 28(1):235–42.
- Sander T, Freyss J, von Korff M, Rufener C. DataWarrior: An Open-Source Program for Chemistry Aware Data Visualization and Analysis. J Chem Inf Model. (2015) 55(2):460–73.
- The PyMOL Molecular Graphics System, Version 1.2r3pre, Schrödinger, LLC.
- Sanner MF. Python: a programming language for software integration and development. J Mol Graph Model. (1999) 17(1): 57–61.
- Trott O, Olson AJ. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. (2009) 31(2): 455-61.
- Molinspiration Cheminformatics free web services, https://www.molinspiration.com. Accessed 14 April 2022.
- Simoben CV, Qaseem A, Moumbock AFA, Telukunta KK, Günther S, Sippl W, et al. Pharmacoinformatic Investigation of Medicinal Plants from East Africa. Mol Inform. (2020) 39(11):2000163.
- Ntie-Kang F, Telukunta KK, Döring K, Simoben CV, A. Moumbock AF, Malange YI, et al. NANPDB: A Resource for Natural Products from Northern African Sources. J Nat Prod. (2017) 80(7):2067–76.
- Krishna Deepak RNV, Abdullah A, Talwar P, Fan H, Ravanan P. Identification of FDA-approved drugs as novel allosteric inhibitors of human executioner caspases. Proteins: Struct Funct and Bioinform. (2018) 86(11):1202–10.
- Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. (2017) 7(1):42717.
- Pires DEV, Blundell TL, Ascher DB. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J Med Chem. (2015) 8(9):4066–72.
- BIOVIA, Dassault Systèmes, Discovery Studio Visualizer, v20.1.0.19295, San Diego: Dassault Systèmes, 2020.
- Schrödinger Release 2022-3: Maestro, Schrödinger, LLC, New York, NY, 2021.
- Chikodili I, Chioma I, Chinwendu N, IfedibaluChukwu E. In-silico study for African plants with possible beta-cell regeneration effect through inhibition of DYRK1A. Sci Phytochem. (2022) 1(1):13–28.
- IfedibaluChukwu Ejiofor I, Chikodili Ekeomodi C, Elomeme S, Ebele Ejiofor M. Therapeutic Inhibitors: Natural Product Options through Computer-Aided Drug Design. In: Shailendra KS, editors. Drug Repurposing - Molecular Aspects and Therapeutic Applications. London: Intechopen; 2022.
- Ejiofor II, Zaman K, Das A. Effect of Extracts of Vernonia Amygdalina in Helminthiasis-A Tropical Neglected Disease. Open Access J Pharm Res. (2017) 1(8):000147.
- Ejiofor II, Zaman K, Das A. Antidiabetic evaluations of different parts of Vernonia amygdalina. IOSR J Pharm Biol Sci. (2017) 12(4):23–8.
- Zothantluanga JH. Ethnopharmacology and phytochemistry-based review on the antimalarial potential of Acacia pennata (L.) Willd. Sci Vis. (2020) 20(4):139–47.
- Ejiofor II, Das A, Mir S, Ali M, Zaman K. Novel phytocompounds from Vernonia amygdalina with antimalarial potentials. Pharmacogn Res. (2020) 12(1):53.
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. (1997) 23(1-3):3–25.
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. (2001) 46(1-3):3–26.
- Lipinski CA. Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov Today Technol. (2004)1(4):337–41.
- BIOVIA Databases | Bioactivity Databases: RTECS. accelrys.com Accessed 15 April 2021.
- Sergeev YV, Dolinska MB, Wingfield PT. Thermodynamic Analysis of Weak Protein Interactions Using Sedimentation Equilibrium. Curr Protoc Protein Sci. (2014) 77(1):20.13.1-20.13.15.
- Du X, Li Y, Xia Y-L, Ai S-M, Liang J, Sang P, et al. Insights into Protein–Ligand Interactions: Mechanisms, Models, and Methods. Int J Mol Sci. (2016) 17(2):144.
- Muthu S, Durairaj B. Molecular docking studies on interaction of Annona muricata compounds with antiapoptotic proteins Bcl-2 and survivin Sky. J Biochem Res. (2016) 5:14–7.
- Ritchie TJ, Ertl P, Lewis R. The graphical representation of ADME-related molecule properties for medicinal chemists. Drug Discov Today. (2011) 16(1-2):65–72.
- Lovering F, Bikker J, Humblet C. Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J Med Chem. (2009) 52(21):6752–6.
- Vijay U, Gupta S, Mathur P, Suravajhala P, Bhatnagar P. Microbial Mutagenicity Assay: Ames Test. Bio Protoc. (2018) 8(6):e2763.
- Claxton LD, de A. Umbuzeiro G, DeMarini DM. The Salmonella Mutagenicity Assay: The Stethoscope of Genetic Toxicology for the 21st Century. Environ Health Perspect. (2010) 118(11):1515–22.
- Stergiopoulos C, Tsopelas F, Valko K. Prediction of hERG inhibition of drug discovery compounds using biomimetic HPLC measurements. ADMET and DMPK. (2021) 9(3):191–207.
- Wu X, Zhang Q, Hu J. QSAR study of the acute toxicity to fathead minnow based on a large dataset. SAR and QSAR in Environ Res. (2016) 27(2):147–64.
- Kalita J, Chetia D, Rudrapal M. Design, Synthesis, Antimalarial Activity and Docking Study of 7-Chloro-4- (2-(substituted benzylidene)hydrazineyl)quinolines. Med Chem. (2020) 16(7):928–7.
- Zothantluanga JH. Molecular Docking Simulation Studies, Toxicity Study, Bioactivity Prediction, and Structure-Activity Relationship Reveals Rutin as a Potential Inhibitor of SARS-CoV-2 3CL pro. J Sci Res. (2021) 65(05):96–104.
- Pasala PK, Abbas Shaik R, Rudrapal M, Khan J, Alaidarous MA, Jagdish Khairnar S, et al. Cerebroprotective effect of Aloe Emodin: In-silico and in-vivo studies. Saudi J Biol Sci. (2021) 29(2): 998–1005.
- Pasala PK, Uppara RK, Rudrapal M, Zothantluanga JH, Umar AK. Silybin phytosome attenuates cerebral ischemia‐reperfusion injury in rats by suppressing oxidative stress and reducing inflammatory response: In-vivo and in-silico approaches. J Biochem Mol Toxicol. (2022) 36(7):e23073.
- Barbhuyan, NU, Tayeng, D, Gogoi, N, Patowary, L, Chetia, D, Barthakur, MS. Design and screening of tetracycline antibiotics: an in-silico approach. Sci Phytochem. (2023) 2(1):8–16.
- Zothantluanga, J, Chetia, D. A beginner’s guide to molecular docking. Sci Phytochem. (2022) 1(2):37-40.
- Sangma, CD, Chetia, D, Borthakur, MS, Patowary, L, Tayeng, D. In-silico design and screening of cephalosporin derivatives for their inhibitory potential against Haemophilus influenza. Sci Phytochem. (2022) 1(2):1–10.
- Patowary, L, Borthakur, MS. Computational studies of Bridelia retusa phytochemicals for the identification of promising molecules with inhibitory potential against the spike protein and papain-like protease of SARS-CoV-2. Sci Phytochem. (2022) 1(1):29–41.
- Pegu, F. Pharmacological activities of bioactive compounds isolated from Acacia pennata (L) Willd.: A comprehensive update and application of in-silico techniques for repurposing. Sci Phytochem. (2022) 1(1):1–12.